

Abstract
Arylsulfatase B (N-acetylgalactosamine-4-sulfatase; ARSB) removes 4-sulfate groups from chondroitin-4-sulfate (C4S) and dermatan sulfate and is required for their degradation. In human prostate stromal and epithelial cells, when ARSB was silenced, C4S, versican, and versican promoter activity increased, and the galectin-3 that co-immunoprecipitated with C4S declined. Galectin-3 silencing inhibited the ARSB-silencing induced increases in versican and versican promoter, due to effects on the AP-1 binding site in the versican promoter. These findings demonstrate for the first time the transcriptional mechanism whereby ARSB can regulate expression of an extracellular matrix proteoglycan with C4S attachments. In addition, following ARSB silencing, C4S that co-immunoprecipitated with versican increased, whereas co-immunoprecipitated EGFR declined, total EGFR increased, and exogenous EGF-induced cell proliferation increased, suggesting profound effects of ARSB on vital cell processes.
Introduction
Arylsulfatase B (ARSB; N-acetylgalactosamine-4-sulfatase) is the enzyme that removes 4-sulfate groups from chondroitin-4-sulfate and dermatan sulfate at the non-reducing end of the sulfated glycosaminoglycan (GAG) chain. ARSB is required for the degradation of these sulfated glycosaminoglycans, as evident in the genetic disorder Mucopolysaccharidosis VI (MPS VI; Maroteaux-Lamy Syndrome) in which ARSB activity is reduced, and sulfated GAGs accumulate throughout the body, leading to marked alteration of normal physiological processes. MPS VI is classified as a lysosomal storage disorder, but recent work has demonstrated that ARSB also localizes on cell membranes of epithelial and endothelial cells [1–5]. In human colonic and prostatic malignancies and in malignant mammary cell lines, ARSB activity was reduced, in association with increased sulfated glycosaminoglycans, largely chondroitin-4-sulfate [2,3,6–8]. In the malignant prostate tissues, higher Gleason scores and recurrent disease were associated with lower ARSB content [3].
These multiple findings suggest an important role for ARSB in oncogenesis, as well as other vital cell processes [9–15]. In studies with hypoxia and ARSB silencing, increased transcription of HIF-1α was associated with increases in nuclear AP-1 and galectin-3 (LGALS3), a galectin with specific affinity for β-galactosidases [16]. Galectins-3,7, and 9 were reported to bind less to more highly sulfated chondroitin sulfates [17], suggesting a potential link between activity of arylsulfatase B and galectin-3 mediated processes. Interaction between galectin-3 and AP-1 on activation of the MUC-2 promoter in colonic epithelial cells had been reported previously [18], and galectin-3 has been recognized as a mediator in prostate oncogenesis [19], but with conflicting results in other malignancies [20].
In this report, the impact of knockdown of ARSB and of galectin-3 on versican promoter activity is addressed. For the first time, we present a transcriptional mechanism by which chondroitin sulfation regulates expression of a proteoglycan with chondroitin sulfate attachments. Both versican and in chondroitin sulfate have been reported to be increased in malignant prostate tissues and to be useful as biomarkers of more aggressive disease [21–23]. Versican is recognized as an important mediator of cell-matrix interactions in mammalian tissues, with both chondroitin sulfate and hyaluronan attachments and with EGF-like repeats at the C-terminus [24,25]. Since the versican promoter has an AP-1 binding site [26], we have probed the effects of ARSB, chondroitin-4-sulfate, and galectin-3, on versican expression. The mechanism, whereby changes in chondroitin sulfation due to decline in ARSB activity affects versican expression, indicates how external signals that affect ARSB activity, including hypoxia and increased chloride, can lead to transcriptional events that affect the composition of the extracellular matrix and stromal-cellular interactions.
RESULTS
Measurements of Arylsulfatase B activity, total sulfated glycosaminoglycans, and chondroitin-4-sulfate
Arylsulfatase B (ARSB) activity was measured in prostate stromal and epithelial cells and in prostate tissue from ARSB-deficient mice and control mice using the exogenous substrate 4-methylumbilleferylsulfate. Baseline activity declined from 140.4 ± 8.4 nmol/mg protein/h to 21.4 ± 0.9 nmol/mg protein/h in the stromal cells (Fig. 1A) and from 110.6 ± 6.8 nmol/mg protein/h to 8.8 ± 0.8 nmol/mg protein/h in the epithelial cells following ARSB silencing by siRNA (Fig. 1B). Control silencing produced no change in activity. Differences were highly significant (p<0.001).
FIGURE 1. Decline in ARSB activity leads to increases in total sulfated glycosaminoglycans and chondroitin-4-sulfate in prostate stromal cells, prostate epithelial cells, and ARSB null mice.

Panel A. In human prostate stromal cells, ARSB activity declined significantly following ARSB silencing (p<0.001). [ARSB=Arylsulfatase B]
Panel B. In human prostate epithelial cells, ARSB activity declined significantly following ARSB silencing (p<0.001). Baseline activity was ~21% less than in the stromal cells.
Panel C. In ARSB null mice, baseline activity was markedly reduced, to 7.0 ± 0.7 nmol/mg protein/h vs. 111.8 ± 11.1 nmol/mg protein/h in the heterozygous mouse prostate tissue (p<0.001, unpaired t-test, two-tailed).
Panel D. Total sulfated GAGs, including chondroitin-4-sulfate, chondroitin-6-sulfate, dermatan sulfate, heparin, heparan sulfate, and keratan sulfate, were significantly increased when ARSB was silenced in the stromal cells, increasing by ~10.7 μg/mg protein (p<0.001).
Panel E. In the epithelial cells, the total sulfated GAGs increased by ~6.2 μg/mg protein when ARSB was silenced (p<0.001).
Panel F. In the mouse prostate tissue, the sulfated GAGs were 5.0 μg/mg higher in the ARSB null mice than in the heterozygous controls (p=0.02, unpaired t-test, two-tailed).
Panel G. In the stromal cells, increase in C4S accounted for most of the increase in the total sulfated GAGs, increasing by ~8.0 μg/mg protein in the stromal cells.
Panel H. Similarly in the epithelial cells, the increase in C4S was ~5.5 μg/mg protein.
Panel I. In the prostate of the ARSB deficient mice, the C4S was ~4.1 μg/mg protein greater than in the heterozygous mouse (p<0.001, unpaired t-test, two-tailed). [ARSB=Arylsulfatase B; GAG=glycosaminoglycans; C4S=chondroitin-4-sulfate]
In the ARSB-null mice, ARSB activity was 7.0 ± 0.7 nmol/mg protein/h, compared to 111.8 ± 11.1 nmol/mg protein/h in the control mouse prostate tissue (p<0.001, unpaired t-test, two-tailed) (n=6) (Fig. 1C). Consistent with the decline in ARSB, sulfated glycosaminoglycans (GAGs) and C4S were significantly increased in the prostate stromal and epithelial cells following ARSB silencing and in the prostate tissue of the ARSB null mice vs. the control (Fig. 1D–1I). In the stromal cells, total sulfated GAGs increased from 17.9 ± 0.7 μg/mg protein to 28.6 ± 1.4 μg/mg protein (p<0.001); in the epithelial cells, sulfated GAGs increased from 18.9 ± 0.6 μg/mg protein to 25.1 ± 1.2 μg/mg protein (p<0.001). The sulfated GAG concentration in the prostate tissue of the ARSB-deficient mice was 21.3 ± 1.8 μg/mg protein, compared to 16.3 ± 1.5 μg/mg protein in age-matched wild-type C57BL/6J controls (p=0.02, unpaired t-test, two-tailed) (Fig. 1F). C4S increased from 7.0 ± 0.3 μg/mg protein in the heterozygous mice to 11.1 ± 0.4 μg/mg protein in the ARSB null mice (p<0.001, unpaired t-test, two-tailed) (Fig. 1I). The increases in the total sulfated GAGs were largely accounted for by increased C4S, amounting to ~8.0 μg/mg protein in the stromal cells, ~5.5 μg/mg protein in the epithelial cells, and ~4.1 μg/mg protein in the mouse prostate when ARSB was defective.
ARSB silencing leads to redistribution of galectin-3 and to decline in galectin-3 that co-immunoprecipitates with C4S
Following ARSB silencing in the prostate stromal and epithelial cells, total cellular and nuclear galectin-3 increased, and galectin-3 in the media declined (p<0.001) (Fig. 2A, 2B). In the stromal cells, nuclear galectin-3 increased to ~twice the baseline, from 7.2 ± 0.6 ng/mg protein to 14.3 ± 0.8 ng/mg protein, and in the epithelial cells, nuclear galectin-3 increased to 2.2 ± 0.2 times the baseline level (p<0.001). In the ARSB null mice, cellular galectin-3 in the prostate tissue was 2.1 ± 0.2 times the level in the control tissue, and nuclear galectin-3 was 1.9 times the level in the control prostate tissue (p<0.001, unpaired t-test, two-tailed) (Fig. 2C). The increases in nuclear galectin-3 accounted for most of the total cellular increase. The mRNA expression of galectin-3 was not changed significantly in either the stromal or epithelial cells following ARSB silencing.
FIGURE 2. Redistribution of galectin-3 and decline in galectin-3 co-immunoprecipitated with C4S following ARSB silencing.
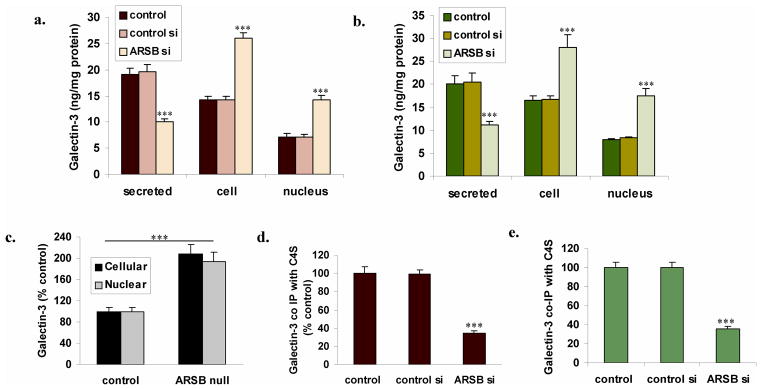
Panel A. In the stromal cells, total cellular and nuclear galectin-3 both increased to twice the baseline level when ARSB was silenced, and galectin-3 in the media declined (p<0.001).
Panel B. Similarly, in the epithelial cells, the total cellular and nuclear galectin-3 increased to more than twice their baseline level when ARSB was silenced, and galectin-3 in the media declined (p<0.001).
Panel C. In the ARSB null mice, the cellular and nuclear galectin-3 in the prostate tissue was ~two times greater than the level in the control mouse prostate tissue (p<0.001, unpaired t-test, two-tailed). The increase in nuclear galectin-3 largely accounted for the total increase.
Panel D. After ARSB silencing, the galectin-3 that co-immunoprecipitated with C4S in the stromal cells declined to 35% of the baseline value. This is consistent with previous observations that galectin-3 binds less to more highly sulfated chondroitin sulfate and enables intracellular signaling by changes in galectin-3 and chondroitin sulfate binding when ARSB activity is reduced (p<0.001).
Panel E. Similarly, in the epithelial cells, the galectin-3 that co-immunoprecipitated with chondroitin sulfate using the C4S antibody declined to 35.5 ± 2.4 % of the baseline value (p<0.001).
In contrast to the increases in total cellular and nuclear galectin-3, the galectin-3 that was bound to C4S in the stromal and epithelial cells declined to ~35% of the baseline value following ARSB silencing (p<0.001) (Fig. 2D,2E), consistent with previous findings [16] that galectin-3 bound less tightly to the more highly sulfated chondroitin sulfate present when ARSB activity was reduced.
ARSB silencing-induced increases in nuclear c-Jun and c-Fos are inhibited by galectin-3 silencing
Effectiveness of galectin-3 silencing was demonstrated in confocal images of galectin-3 in the prostate epithelial cells, in which galectin-3 was not silenced (Fig. 3A) or was silenced by siRNA (Fig. 3B). Residual galectin-3 present after silencing was localized in the perinuclear area. ARSB silencing increased nuclear c-Jun and c-Fos, as measured by binding to the AP-1 consensus oligonucleotide adherent to the wells of an ELISA plate. In the stromal cells, ARSB silencing produced an increase of 3.53 ± 0.31 times the baseline in nuclear c-Jun and an increase of 3.87 ± 0.04 times the baseline in nuclear c-Fos (p<0.001) (Fig. 3C). Similarly, in the prostate epithelial cells, knockdown of ARSB produced an increase in nuclear c-Jun to 3.60 ± 0.05 times the baseline and increase in nuclear c-Fos to 3.47 ± 0.36 times the baseline (p<0.001) (Fig. 3D). These increases were completely inhibited when galectin-3 was silenced in combination with ARSB silencing (Fig. 3C, 3D). Increases in nuclear AP-1 were also present in the prostate tissue of the ARSB null mice, compared to control prostate tissue (Fig. 3E). c-Jun increased to 3.13 ± 0.22 times the baseline, and c-Fos increased to 3.51 ± 0.23 times the baseline value (p<0.001, unpaired t-test, two-tailed).
FIGURE 3. Increases in nuclear c-Jun and c-Fos following ARSB knockdown are inhibited by galectin-3 silencing.

Panel A. Confocal images demonstrate that galectin-3 is abundant in the cytoplasm and perinuclear region in the prostate epithelial cells following ARSB silencing.
Panel B. Galectin-3 silencing effectively reduces the galectin-3 present in the prostate epithelial cells. The remaining galectin-3 is localized near the nuclear chromatin.
Panel C. Nuclear c-Jun and c-Fos were measured by binding to the AP-1 consensus oligonucleotide coated wells of an ELISA plate. Following ARSB silencing, in the prostate stromal cells, c-Jun increased to 3.53 ± 0.31 times the baseline and c-Fos increased to 3.87 ± 0.04 times the baseline. Galectin-3 silencing in combination with ARSB silencing inhibited these increases and the decline is highly significant (p<0.001).
Panel D. In the prostate epithelial cells, ARSB knockdown led to similar increases with nuclear c-Jun increasing to 3.60 ± 0.05 times baseline and nuclear c-Fos increasing to 3.47 ± 0.36 times baseline. Again, galectin-3 siRNA in combination with ARSB siRNA completely inhibited these increases. The differences following ARSB silencing are highly significant (p<0.001), vs. the control silencing and the Galectin-3 silencing alone or in combination with the ARSB silencing.
Panel E. In the prostate tissue of the ARSB null mice, similar increases in activated c-jun and c-fos were detected. c-Jun increased to over three times baseline, and c-Fos increased to ~3.51 times the baseline value. These differences are highly significant (p<0.001, unpaired t-test, two-tailed).
ARSB silencing-induced increased in versican is inhibited by galectin-3 silencing and by AP-1 inhibitors
Confocal images demonstrated the increase in abundance of versican protein in the prostate stromal cells following ARSB silencing, compared to control silencing (Fig. 4A–4D). Marked increase in versican fluorescent immunostaining (red) was apparent, including at the cell membrane. Corresponding to these images, versican protein measured by ELISA increased from baseline of 121.0 ± 5.0 ng/mg protein to 287.5 ± 20.7 ng/mg protein in the prostate stromal cells (p<0.001) (Fig. 4E). In the prostate epithelial cells, versican increased from 141.6 ± 12.1 to 319.1 ± 11.9 ng/mg protein (p<0.001) (Fig. 4F). In the ARSB null mice, prostate versican content was 217.1 ± 32.2 ng/mg protein, compared to 125.9 ± 7.3 ng/mg protein in the control mouse prostate tissue (p<0.01, unpaired t-test, two-tailed) (Fig. 4G).
FIGURE 4. Versican is increased by ARSB silencing, and this increase is inhibited by galectin-3 silencing and by AP-1 inhibitors.

Panels A,B,C,D. Prostate stromal cells were grown in transwells, and cells were stained following treatment with control siRNA or ARSB siRNA. Versican was immunostained with mouse monoclonal antibody and goat anti-mouse Alexa Fluor 594 (Invitrogen), and visualized. Versican immunostaining is faint in the control prostate cells (A), but intense red staining is present following ARSB silencing (B). Merged images with staining for β-actin (green) and DNA (blue) demonstrate prominent versican immunostaining. IgG control showed no red staining (not shown).
Panel E. Consistent with the images above, versican protein increased from baseline of 121.0 ± 5.0 ng/mg protein to 287.5 ± 20.7 ng/mg protein in the prostate stromal cells when ARSB was silenced (p<0.001).
Panel F. Similarly, in the prostate epithelial cells, versican increased from baseline of 141.6 ± 12.1 to 319.1 ± 11.9 ng/mg protein when ARSB was silenced (p<0.001).
Panel G. In the ARSB deficient mice, the prostate versican protein was 217.1 ± 32.2 ng/mg protein, significantly more than in the control mouse prostate (125.9 ± 7.3 ng/mg protein; p<0.01, unpaired t-test, two-tailed).
Panel H. When ARSB was silenced in the prostate stromal cells, versican mRNA expression increased more than two-fold (p<0.001). This increase was completely inhibited when galectin-3 was also silenced.
Panels I, J. The combination of galectin-3 silencing and ARSB silencing nullified the increase in ARSB protein that followed ARSB silencing in the prostate stromal and epithelial cells (p<0.001). These findings demonstrate that the increased expression of versican following ARSB silencing requires galectin-3.
Panel K. When prostate stromal cells were exposed to inhibitors of AP-1, identified as I1 and I2, the ARSB-knockdown induced increase in versican was reversed (p<0.001). I1 is a c-Jun mimetic peptide that impairs binding of JNK to c-Jun. I2 is an oligonucleotide-binding inhibitor that competes with c-Fos for the AP-1 oligonucleotide binding site. he inhibitors I1 and I2 inhibited the ARSB-knockdown induced increase in versican protein in the prostate stromal cells (p<0.001).
Following ARSB silencing, versican mRNA expression in the stromal cells increased to 2.13 ± 0.28 times the baseline (p<0.001), and this increase was inhibited by combined silencing with galectin-3 siRNA (Fig. 4H). Consistent with this effect, galectin-3 silencing inhibited the increases in versican protein produced by ARSB silencing in the stromal and epithelial cells (p<0.001) (Fig. 4I, 4J). These findings indicate that the increased expression of versican following ARSB silencing required galectin-3.
Prostate stromal cells were also exposed to inhibitors of AP-1, identified as I1 and I2 [27,28]. I1 is a c-Jun mimetic peptide, composed of residues 33–57 of the JNK binding domain of human c-Jun, and impairs binding of JNK to c-Jun. I2 is an oligonucleotide-binding inhibitor that competes with c-Fos for the AP-1 oligonucleotide binding site. Both I1 and I2 completely inhibited the ARSB-knockdown induced increase in versican protein in the prostate stromal cells (p<0.001).
ARSB silencing-induced increases in versican and versican promoter activity are inhibited by galectin-3 silencing and by AP-1 inhibitors
To explain the effects of ARSB silencing and galectin-3 silencing on the increase in versican expression, we examined changes in versican promoter activity by measurement of relative luciferase units following transfection with the versican promoter construct in a Renilla luciferase reporter. When ARSB was silenced in the prostate stromal cells, versican promoter activity increased to 3.14 times the baseline (p<0.001) (Fig. 5A). Positive actin promoter control increased 4-fold over baseline (for control cells: 42,769 ± 3251), but was unaffected by ARSB silencing, galectin-3 silencing, or combined ARSB and galectin-3 silencing. Negative transfection control readings were consistently low (for control cells: 3179 ± 282) and unaffected by silencing of ARSB or galectin-3. The combination of ARSB silencing and galectin-3 silencing inhibited the increase in versican promoter activity induced by ARSB silencing. In the prostate epithelial cells, versican promoter activity increased to 2.70 times the baseline activity when ARSB was silenced (p<0.001) (Fig. 5B). Again, this effect was completely reversed when galectin-3 was also silenced. These findings demonstrated that the effect of ARSB silencing on versican promoter activation was dependent upon galectin-3, and support a mechanism whereby changes in C4S caused by decline in ARSB activity can produce transcriptional effects.
FIGURE 5. ARSB silencing increases versican promoter activity, and this increase is blocked by galectin-3 silencing and by AP-1 inhibitors.


Panel A. To further assess the impact of ARSB silencing and galectin-3 silencing on versican expression, versican promoter activity was determined following ARSB silencing, galectin-3 silencing, and the combination of ARSB and galectin-3 silencing. In the prostate stromal cells, ARSB silencing increased versican promoter activity to 3.14 times the baseline (p<0.001). This increase was completely inhibited when both ARSB and galectin-3 were silenced. Negative vector control demonstrates no changes; positive actin control is not shown. No further decline in versican promoter activity occurred when ARSB and galectin-3 were both silenced and the inhibitors were present.
Panel B. In the prostate epithelial cells, when ARSB was silenced, versican promoter activity increased to 2.70 times the baseline activity (p<0.001). This increase was completely abrogated by the combination of ARSB and galectin-3 silencing, demonstrating dependence of the ARSB knockdown-induced effect on the versican promoter on galectin-3, and consistent with the observed change in galectin-3 binding to C4S when ARSB was silenced. [ARSB=Arylsulfatase b; C4S=chondroitin-4-sulfate]
Panel C. Chromatin immunoprecitiation (CHiP) assay was performed with c-Fos antibody following ARSB silencing and in the presence of c-Fos oligonucleotide binding inhibitor (I2). Epithelial and stromal cells showed significant increases in % DNA captured following ARSB silencing. Panel D. CHiP assay with galectin-3 antibody also demonstrated increase following ARSB silencing that was inhbited in the presence of I2.
Panel E. Increased intensity of the chromatin bands is apparent following immunoprecipitation in the presence of c-Fos in the stromal and epithelial cells.
Panel F. Similarly, immunoprecipitation with galectin-3 demonstrates increased band intensity following ARSB silencing.
Panel G. c-Fos from the nuclear fraction of stromal cells bound with to the AP-1 binding sequence coated onto the wells of an ELISA plate. Anti-rabbit c-Fos and secondary goat-anti-rabbit-IgG-FITC were used to detect the c-Fos and were measured by fluorescence. Following ARSB silencing, the bound c-Fos was 2.25 and 2.30 times the baseline level in the stromal cells and in the epithelial cells, respectively. The I2 inhibitor completely blocked the binding.
Panel H. Galectin-3 binding to the AP-1 oligonucleotide consensus sequence was also increased following ARSB silencing and inhibited by I2. When galectin-3 binding was detected by a mouse anti-galectin antibody and goat anti-mouse-IgG-Rhodamine Red™X, the binding was shown to have increased to 4.3 and 4.3 times the baseline, in the stromal and epithelial cells, respectively.
Panel I. In the ARSB null mouse prostate tissue, the nuclear c-Fos and galectin-3 were both increased. C-Fos increased to 2.51 times the baseline and galectin-3 to 2.54 times the baseline (p=0.0002, p<0.0001, unpaired t-test, two-tailed).
Panel J. Schematic diagram shows reduced binding of galectin-3 to more sulfated chondroitin-4-sulfate (C4S) that is present when ARSB is diminished. This is associated with increased nuclear galectin-3 and increased association of galectin-3 with c-Jun and c-Fos, leading to increased transcription of versican. [ARSB=arylsulfatase B; blue box = sulfate group at non-reducing end of C4S].
Exposure of the prostate stromal and epithelial cells to the c-Jun inhibitory peptide (I1) or the oligonucleotide inhibitor of c-Fos binding to the AP-1 consensus sequence (I2), completely inhibited the ARSB-induced increases in versican promoter activity and versican protein expression in the prostate stromal (Fig. 5A) (p<0.001) and epithelial cells (Fig. 5B) (p<0.001). No further decline in versican promoter activity occurred when ARSB and galectin-3 were both silenced and the inhibitors were present. These findings are consistent with mediation of ARSB effects on versican transcription through the AP-1 binding site.
Chromatin immunoprecipitation (ChIP) assay was used to demonstrate binding of c-Fos and of Galectin-3 to the versican promoter. Sheared chromatin from control, control-silenced, and ARSB-silenced prostate stromal and epithelial cells was incubated with c-Fos antibody, Galectin-3 antibody, or IgG control, and protein-DNA complexes were immunoprecipitated. DNA was isolated and amplified to quantify abundance of the versican promoter. QPCR demonstrated marked increase in the versican promoter precipitated by c-Fos (Fig. 5C, 5E) and by Galectin-3 (Fig. 5D, 5F) following ARSB silencing in both the stromal and epithelial cells (p<0.001). When cells were treated with the c-Fos oligonucleotide binding inhibitor (I2), the DNA isolated was negligible (Fig. 5C-5F). The marked increase in versican promoter oligonucleotide band intensity when ARSB was silenced was demonstrated on agarose gels (Fig. 5E, 5F), and band intensity was markedly diminished in the presence of I2.
A modified oligonucleotide ELISA was performed, in which microplate wells were coated with the AP-1 consensus oligonucleotide and fluorescent secondary antibodies were used to quantify c-Fos and galectin-3 binding. Marked increases followed ARSB silencing in the stromal (Fig. 5G) and epithelial cells (Fig. 5H) and in the ARSB null mice (Fig. 5I). A schematic illustration demonstrates the effect of ARSB reduction on galectin-3 binding to C4S, with subsequent increase in nuclear activation of AP-1 and increased expression of versican (Fig. 5J).
Following ARSB silencing, C4S co-immunoprecipitated with versican increases, whereas EGFR that co-immunoprecipitates with versican declines
Versican in the prostate stromal and epithelial cells was quantified by ELISA and equal amounts of versican were immunoprecipitated by versican antibodies. The versican-associated C4S and EGFR were measured by Blyscan assay and by ELISA, respectively. C4S co-immunoprecipitated with versican following ARSB silencing increased from 184 ± 3 ng/ug versican to 448 ± 25 ng/ug versican (Fig. 6A). In contrast, total EGFR (phosphorylated and unphosphorylated ErbB1 + EGFR) that co-immunoprecipitated with versican declined from 98.8 ± 6.6 ng/μg versican to 16.6 ± 0.86 ng/μg versican (Fig. 6B). Although EGFR associated with versican declined, the total EGFR in the stromal (Fig. 6C) and epithelial (Fig. 6D) cells increased markedly (p<0.001). These differences suggest that increases in C4S on the G2 domain of versican following ARSB decline may sterically inhibit the binding of the EGF-like motifs on the G3 domain with EGFR. This may lead to reduced binding of the EGF-like repeats of versican with EGFR binding, even though cellular EGFR is increased.
FIGURE 6. Following ARSB silencing, C4S co-immunoprecipitated with versican increases, whereas EGFR that co-immunoprecipitates with versican declines and total EGFR increases.

Panel A. The amount of C4S that co-immunoprecipitates with versican increased to ~2.5 times the baseline level in the prostate stromal and epithelial cells when ARSB was silenced (p<0.001).
Panel B. In contrast, the EGFR that co-immunoprecipitated with versican declined to about 17% of the baseline value (p<0.001), with higher levels in the epithelial than the stromal cells.
Panels C,D. The total EGFR was almost three times greater in both the stromal and epithelial cells following ARSB silencing (p<0.001), although less was associated with the versican.
Panel E. The prostate epithelial cells were deprived of EGF, then challenged with exogenous EGF (10 ng/ml × 24 h, and the BrdU incorporation was detected. BrdU incorporation was significantly greater in the ARSB silenced cells than in the control or control silenced cells, increasing by 29% (p<0.001; n=6).
To test if the reduced binding of versican EGF-like repeats with EGFR might permit more effect of exogenous EGF, the epithelial cells were challenged with exogenous EGF (10 ng/ml × 24 h). The 5-bromo-2′-deoxyuridine (BrdU) incorporation was 29% greater in the ARSB silenced cells than in the control cells (p<0.001) (Fig. 6E), consistent with an impact of ARSB knockdown on cell proliferation.
DISCUSSION
These results indicate demonstrate that ARSB can modify the expression of a chondroitin-4-sulfate containing proteoglycan, thereby potentially regulating chondroitin-4-sulfate localization, versican functionality, and cell proliferation. Versican is a critically important proteoglycan in the extracellular matrix, so changes in its expression have the potential to affect many vital cell-matrix interactions. Versican interacts with multiple epithelial cell surface receptors and modulates signaling pathways, including the EGF-EGFR pathway. Two epidermal growth factor (EGF)-like repeats are located in the G3 domain of versican, and the interaction of the versican EGF-like repeats with the epithelial cell EGFR has been reported to affect EGFR signaling and to influence cell growth and invasiveness [29–31]. Both versican and chondroitin sulfate have been identified as potential biomarkers for prostate cancer progression [21–23]. The findings in this report demonstrate that ARSB can regulate these processes in prostate cells and suggest a mechanism whereby decline in ARSB activity can modulate cell proliferation.
The study findings do not exclude involvement of other C4S-containing proteoglycans, either lecticans or small-leucine rich proteoglycans, in the cellular responses to reduced ARSB. Similar changes might occur with other proteoglycans with EGF-like motifs and chondroitin sulfate attachments. The chondroitin sulfate attachments on the G2 region of versican likely include C6S chains which can contribute to an inhibitory effect on the interaction of the versican’s EGF-like regions with EGFR. We do not have precise measurements of C6S present in versican, but overall, in the cells and tissue, the measured increases in C4S account for 89.3% of the total increase in sulfated GAGs in the epithelial cells, 74% in the stromal cells, and 82% in the ARSB-deficient mice compared to WT. The remainder includes increases in C6S, as well as heparin, heparan sulfate, dermatan sulfate, and keratan sulfate. Additional studies are required to identify the impact of decline in ARSB on other proteoglycans and contribution of C6S to the observed effects.
ARSB activity can be affected by several environmental factors, including oxygen, chloride, phosphate, sulfate, vanadate, and by post-translational modification by the formylglycine generating enzyme which requires oxygen [12,15,16,32–36]. ARSB may act as a cellular rheostat, responding to multiple diverse extracellular cues, then mediating transcriptional events, through diminished galectin-3 binding to more highly sulfated chondroitin-4-sulfate. Nuclear galectin-3 interacts with heterogeneous nuclear ribonucleoprotein [37,38], suggesting the potential for multiple effects on transcription.
This report extends considerations about how changes in sulfation can impact upon transcriptional events in mammalian cells, but the cellular metabolism of sulfate requires more attention. Recombinant human ARSB is currently safely and effectively used in replacement therapy for the genetic disorder Mucopolysaccharidosis VI [39], and intervention with ARSB supplementation may potentially be of benefit in other disorders.
MATERIALS AND METHODS
Human prostate cell lines and mouse prostate tissue
Human prostate stromal cells (ATCC®: CRL-2850™) and prostate epithelial cells (ATCC®: CRL-2854™) were obtained and grown under the recommended conditions. Cells were grown to ~70–80% confluency, treated as indicated below, and harvested by scraping. Heterozygous arylsulfatase B deficient mice were obtained (Strain 005598; Jackson Laboratories, Bar Harbor, Maine) and bred [40]. Genotyping was performed to detect homozygous or heterozygous mutation or wild-type ARSB. The mutation in ARSB (NM_009712.3) is a G to T mutation at residue 94,560,057 (UCSC database) in exon 2 [39], leading to truncation and reduced activity. The TaqMan Sample-to-SNP kit (Life Technologies) and DreamTaq Green PCR Master Mix (2X) (Fermentas) were used to extract DNA from mouse tail specimens and to perform PCR. The ARSB primers used for PCR were determined using Primer3 software [41]. They were: (left forward): 5′ GCA GTG GGT TTG GAA TCC T 3′ and (right reverse): 5′ ACT GTG TTC AGG TTT CTG GTC A 3′. Direct sequencing was performed in the DNA facility of the University of Illinois at Chicago (UIC), confirming the presence of G to T mutation at position 94,560,057 in exon 2 of the ARSB mice homozygous for the mutation. No changes in arylsulfatase A, galactose-6-sulfatase, or steroid sulfatase activity were detected in the ARSB null mice.
All animal procedures were approved by the Animal Care Committee of UIC. ARSB defective mice and age- and gender-matched C57BL/6J control mice were euthanized by carbon dioxide inhalation and cervical dislocation, and the prostate gland was isolated, excised, and promptly frozen at −80°C.
N-acetylgalactosamine-4-sulfatase (arylsulfatase B; ARSB), arylsulfatase A, and galactose-6-sulfatase activity assays
Determinations of sulfatase activity were performed in prostate stromal and epithelial cells, grown under the recommended conditions, and in ARSB-deficient and control mouse prostate tissue. Measurements were made from cell homogenates of cultures grown to 70–80% confluence. Similar methods were used for each enzyme assay in the different cell types and mouse tissue. Assays were performed using at least triplicate biological samples with technical replicates of each measurement.
Arylsulfatase B (ARSB) measurements were performed using a fluorometric assay, following a standard protocol with 20 μl of homogenate and 80 μl of assay buffer (0.05 M Na acetate buffer, pH 5.6) with 100 μl of substrate (5mM 4-MUS in assay buffer) in a black microplate, as previously reported [8,12]. To verify that silencing of ARSB was not associated with generalized reduction of sulfatase activity, galactose-6-sulfatase (GALNS), arylsulfatase A (ARSA), and steroid sulfatase activities were also determined, as previously [8,12].
Measurement of total sulfated glycosaminoglycans (sGAG) and chondroitin-4-sulfate
Total sulfated glycosaminoglycans (sGAG) measured include chondroitin 4-sulfate (C4S), chondroitin 6-sulfate, keratan sulfate, dermatan sulfate, heparan sulfate, and heparin. They were measured in prostate cells and prostate tissue from ARSB null mice and control mice by sulfated GAG assay (Blyscan™, Biocolor Ltd, Newtownabbey, Northern Ireland) as previously [1,6]. Disaccharide analysis confirmed the high selectivity of this antibody for C4S (89.1 ± 1.9% C4S disaccharides), and cross-reactivity of the anti-C4S with CS-E or C6S was previously excluded [6].
ARSB silencing and galectin-3-silencing by siRNA
Small interfering (si) RNAs to silence human ARSB and human galectin-3 were obtained commercially (Qiagen, Valencia, CA). The siRNA sequences for ARSB (NM_000046) silencing were: sense 5′-GGGUAUGGUCUCUAGGCA - 3′ and antisense: 5′-UUGCCUAGAGACCAUACCC - 3′. The sequence of the DNA template for human galectin-3 silencing (Hs_LGALS3_9) was: 5′ - ATGATGTTGCCTTCCACTTTA - 3′. Cells were grown to ~60% confluence, then silenced by adding 0.6 μl of 20 μM siRNA (150 ng), mixed with 100 μl of serum-free medium and 12 μl of HiPerfect Transfection Reagent (Qiagen), as previously [16]. Galectin-3 silencing was confirmed by galectin-3 ELISA, with galectin-3 declining ~ 90%.
Versican and galectin-3 mRNA expression
Primers for versican and galectin-3 QPCR were determined using Primer3 software [41]. mRNA expression was normalized by comparison to β-actin expression. Primers for versican were: (left forward) 5′ - CCA CTC TGT TTT CTC CCC ATT - 3′ and (right reverse) 5′-ATC CCT TTG TGC CCT TTT TC -3′. Primers for galectin-3 were: (left forward) 5′ - GGC CAC TGA TTG TGC CTT AT -3′ and (right reverse) 5′ - AAG CGT GGG TTA AAG TGG AAG -3′. Fold-changes following ARSB silencing (treated) and control silencing (control) were quantified using the differences between the cycle thresholds (Ct) for versican or galectin-3 and actin and the formulae: ; Δ1 =Ct [control or control si] [versican or galectin-3] − Ct [control or control si] actin and Δ2 =Ct treated [versican or galectin-3] − Ct treated actin.
Quantification of versican, galectin-3, and epidermal growth factor receptor by ELISA
Mouse and human versican and human galectin-3 proteins were determined using commercial sandwich ELISA kits following the recommended procedures, as previously described [3,16]. Total human EGFR (phosphorylated and unphosphorylated EGFR and ErbB1; R&D, Minneapolis, MN) was also quantified by ELISA [3]. Galectin-3 was measured by ELISA (R&D, Minneapolis, MN) in control and treated cell and nuclear preparations and mouse prostate tissue, and following immunoprecipitation of the prostate cells with C4S antibody (Abnova).
Oligonucleotide-based ELISA to detect nuclear c-jun and c-fos
Oligonucleotide binding assay (TransAM Kit, Active Motif, Carlsbad, CA) was used to detect nuclear c-Jun and c-Fos in the prostate stromal and epithelial cells, following control silencing, ARSB silencing, galectin-3 silencing, combined ARSB and galectin-3 silencing, and in the untreated control cells and in ARSB null and control mouse tissue. Nuclear extracts were prepared using a nuclear extract preparation kit (Active Motif), and were added to the wells of a 96-well microtiter plate, pre-coated with the AP-1 consensus oligonucleotide sequence: 5′-TGAGTCA-3′, as previously described [42]. Sample values were normalized by total cell protein and expressed as percent of untreated control.
Versican promoter activity by luciferase reporter
Human versican promoter construct in a Renilla reniformis luciferase reporter gene (Ren SP) vector was used to determine differences in versican promoter activity following ARSB silencing, control silencing, galectin-3 silencing, and combined ARSB and galectin-3 silencing in the prostate stromal and epithelial cells (LightSwitch Assay, SwitchGear Genomics, Menlo Park, CA). The β-actin promoter (GoClone™) construct was a positive control and a scrambled sequence (R01) was a negative control, both with Renilla luciferase reporters; these were used to determine effectiveness of transcription and specificity of reactions. Transfections were performed with cells at 70% confluence, following silencing for 24 h (as above), using FuGENE HD transfection reagent with proprietary LightSwitch Assay Reagent (SwitchGear). Measurements of luminescence were made after incubation for 24 hours, and compared among the different cell preparations. Versican promoter activity was also determined by this method following inhibition of AP-1 by specific inhibitors (see below). Luminescence was read at 480 nm in a microplate reader (BMG).
Inhibition of AP-1 activation by c-Jun and c-Fos inhibitors
To further assess the role of the AP-1 binding site on versican expression and versican promoter activation, prostate stromal and epithelial cell lysates were exposed to a cell permeable c-Jun mimetic peptide (ILKQSMTLNLADPVGSLKPHLRAKN; R & D; called I1) that comprises residues 33–57 of the human c-Jun binding domain (δ) and thereby prevents JNK/c-Jun interaction and phosphorylation of c-Jun [27]. Effects of exposure to the c-Jun mimetic peptide (400 μM × 2h pre-treatment) on versican promoter activity and versican expression were determined, using the procedures described above. In addition, prostate cells were treated with SR11302 (Santa Cruz Biotechnology; called I2), a retinoid that inhibits AP-1 transcription factor activity by blocking binding to the AP-1 consensus sequence [28]. Effects of exposure to SR11302 (5 μM × 2h pre-treatment) on versican promoter activity and on versican expression were determined as above. The impact of ARSB silencing, galectin-3 silencing, and the combination of ARSB and galectin-3 silencing on versican promoter activity and versican protein was determined following exposure to each inhibitor.
c-Fos and Galectin-3 binding to versican promoter assessed by chromatin immunoprecipitation (ChIP) assay
For chromatin immunoprecipitation (ChIP) assay, IgG control, control silenced, ARSB-silenced, and ARSB-silenced and exposed to c-Fos oligonucleotide binding inhibitor (I2), prostate stromal and epithelial cells were fixed with 1% formaldehyde for 10 min at room temperature. This was followed by shearing of chromatin by sonication (ChIP Assay, Active Motif, Carlsbad, CA). Sheared DNA was incubated with rabbit polyclonal anti-galectin-3 (sc-20157, SCBT, Santa Cruz, CA) and anti-c-Fos (Active Motif) antibodies for 1 h, as well as with IgG control. Protein–DNA complexes were precipitated by protein A/G-coupled magnetic beads. DNA was purified from the immunoprecipitated complexes by reversal of cross-linking and followed by proteinase K treatment. Then, real-time RT-PCR was performed using Brilliant SYBR Green QRT-PCR master mix (Stratagene, La Jolla, CA) and Mx3000 (Stratagene) to amplify the versican promoter. The primers for the versican promoter (forward: 5′-ACCTCTTGGCGTTTCTTCCT-3′ and reverse: 5′-CTCCTTTCCCCTAACCCAGA-3′) encompassed the putative AP-1 binding element. Band intensity was compared among the ARSB silenced, ARSB silenced and inhibitor treated, control silenced, and IgG control samples on a 1.5% agarose gel.
Fluorometric Binding Assay to detect c-Fos and Galectin-3 binding to versican promoter
Nuclear extracts were prepared from control, control silenced, ARSB silenced, and control and ARSB silenced prostate epithelial and stromal cells in the presence of an inhibitor of c-fos oligonucleotide binding (I2) [28] using a nuclear extraction kit (Active Motif). Nuclear Galectin-3 and c-Fos bound to the AP-1 consensus sequence in the versican promoter were detected by fluorescence using a modified AP-1 ELISA (Active Motif). The wells were pre-coated with AP-1 consensus sequence oligonucleotide (5′-TGAGTCA-3′). Nuclear extracts from ARSB silenced, ARSB silenced with I2, control silenced, and control samples were added to the coated wells and incubated for one hour. The oligonucleotide captured the c-Fos and Galectin-3 molecules in the samples. Bound c-Fos and Galectin-3 molecules were then detected by anti-rabbit c-Fos (Active Motif) and anti-mouse galectin-3 (SCBT) antibodies and quantified by secondary goat-anti-rabbit-IgG-FITC and goat anti-mouse-IgG-Rhodamine Red™X secondary antibodies. The fluorescence was measured in a plate reader (FLUOstar, BMG Labtech, Cary NC) at 485/520 nm and 540/620 nm for FITC and Rhodamine Red™X respectively.
Versican and galectin-3 confocal imaging
Prostate stromal and epithelial cells were grown in compartment slides to 70–80% confluence. Cells were treated with ARSB siRNA or control siRNA or control IgG antibody for 24 hours. Cells were washed once in 1x PBS containing 1 mM calcium chloride (pH 7.4), fixed for 1.5 hours with 2% paraformaldehyde, then permeabilized with 0.08% saponin. Preparations were washed with PBS, blocked in 5% normal horse serum (KPL, Inc., Gaithersburg, MD) incubated overnight with goat polyclonal versican antibody (AF3054, R&D) or rabbit polyclonal galectin-3 antibody (sc-20157, SCBT) at 4°C, then washed and stained with Alex Fluor® 594 rabbit anti-goat IgG (H+L) or Alexa Fluor 488 goat anti-rabbit IgG (H+L) (1:100, Invitrogen). Some preparations were exposed for one hour to Alexa Fluor® 488 phalloidin (Invitrogen) diluted 1:40 to stain actin, and slides were coverslipped using DAPI-mounting medium (Vectashield®, Vector Laboratories, Inc., Burlingame, CA) for nuclear staining. Preparations were washed thoroughly, mounted, and observed using Zeiss laser scanning confocal microscope LSM 710 with ZEN software. The fluorochromes were scanned, and the collected images were exported with Zeiss LSM Image Browser software as TIFF files for analysis and reproduction.
Immunoprecipitation of versican and measurements of total EGFR and chondroitin-4-sulfate co-Immunoprecipitated with versican
Versican was measured by ELISA in the immunoprecipitates and equal amounts were used for measuring C4S and EGFR. Total EGFR co-immunoprecipitated with versican antibody (sc-80449, SCBT) was assessed by EGFR ELISA, and C4S co-immunoprecipitated with versican was quantified by Blyscan assay, as above.
5-Bromo-2′-doxyuridine(BrdU) incorporation
BrdU incorporation was measured by a commercial BrdU cell proliferation assay kit (Cell Signaling) following the recommended procedures, as previously [43].
Statistics
Experiments were performed with 3–6 independent biological samples with technical replicates of each sample. Statistical significance of differences between controls and experimental samples was determined by one-way ANOVA with Tukey-Kramer post-test for multiple determinations, unless stated otherwise, with p≤0.05 as significant. In the figures, *** represents p≤0.001, ** represents p≤ 0.01, and * is for p≤ 0.05.
Highlights.
Arylsulfatase B (ARSB) regulates transcription of versican by galectin-3 and AP-1.
Galectin-3 binds less to chondroitin-4-sulfate (C4S) when ARSB is silenced.
Versican’s chondroitin-4-sulfate increases when ARSB is silenced.
Versican’s EGFR declines when ARSB is silenced.
Exogenous EGF leads to increased BrdU incorporation when ARSB is silenced.
Acknowledgments
The authors acknowledge the contribution of Robert Linhardt, Ph.D. and Dr. Bo Yang, who performed the disaccharide analysis.
Footnotes
Conflict of Interest: The authors have no conflict of interest with the content of this manuscript.
References
- 1.Bhattacharyya S, Solakyildirim K, Zhang Z, Linhardt RJ, Tobacman JK. Cell-bound IL-8 increases in bronchial epithelial cells following Arylsulfatase B silencing. Am J Respir Cell Mol Biol. 2010;42:51–61. doi: 10.1165/rcmb.2008-0482OC. [DOI] [PubMed] [Google Scholar]
- 2.Prabhu SV, Bhattacharyya S, Guzman-Hartman G, Macias V, Kajdacsy-Balla A, Tobacman JK. Extra-lysosomal localization of arylsulfatase B in human colonic epithelium. J Histochem Cytochem. 2011;59:328–335. doi: 10.1369/0022155410395511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feferman L, Bhattacharyya S, Deaton R, Gann P, Guzman G, Kajdacsy-Balla A, Tobacman JK. Potential role of Arylsulfatase B as a biomarker for prostate cancer. Prostate Cancer PD. 2013 doi: 10.1038/pcan.2013.18. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitsunaga-Nakatsubo K, Kusunoki S, Kawakami H, Akasaka K, Akimoto Y. Cell-surface arylsulfatase A and B on sinusoidal endothelial cells, hepatocytes, and Kupffer cells in mammalian livers. Med Mol Morphol. 2009;42:63–69. doi: 10.1007/s00795-009-0447-x. [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharyya S, Solakyildirim K, Zhang Z, Linhardt RJ, Tobacman J. Chloroquine reduces arylsulfatase B activity and increases chondroitin 4-sulfate: Implications for mechanisms of action and resistance. Malaria J. 2009;8:303. doi: 10.1186/1475-2875-8-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhattacharyya S, Kotlo K, Shukla S, Danziger RS, Tobacman JK. Distinct effects of N-acetylgalactosamine-4-sulfatase and galactose-6-sulfatase expression on chondroitin sulfate. J Biol Chem. 2008;283:9523–9530. doi: 10.1074/jbc.M707967200. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharyya S, Tobacman JK. Arylsulfatase B regulates colonic epithelial cell migration by effects on MMP9 expression and RhoA activation. Clin Exp Metastasis. 2009;26:535–545. doi: 10.1007/s10585-009-9253-z. [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharyya S, Tobacman JK. Steroid sulfatase, arylsulfatases A and B, galactose 6-sulfatase, and iduronate sulfatase in mammary cells and effects of sulfated and non-sulfated estrogens on sulfatase activity. J Steroid Biochem Mol Biol. 2007;103:20–34. doi: 10.1016/j.jsbmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Katagiri Y, McCann TE, Unsworth E, Goldsmith P, Yu ZX, Tan F, Santiago L, Mills EM, Wang Y, et al. Chondroitin-4-sulfation negatively regulates axonal guidance and growth. J Cell Sci. 2008;121:3083–3091. doi: 10.1242/jcs.032649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoo M, Khaled M, Giubbs KM, Kim J, Kowalewski B, Dierks T, Schachner M. Arylsulfatase B improves locomotor function after mouse spinal cord injury. PLoS One. 2013;8(3):e57415. doi: 10.1371/journal.pone.0057415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrero GB, Pagliardini S, Veljkovic A, Porta F, Bena C, Tardivo I, Restagno G, Silengo MC, Bignamini E. In vivo specific reduction of arylsulfatase B enzymatic activity in children with cystic fibrosis. Mol Genet Metab. 2008;94:39. doi: 10.1016/j.ymgme.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 12.Bhattacharyya S, Look D, Tobacman JK. Increased arylsulfatase B activity in cystic fibrosis cells following correction of CFTR. Clin Chim Acta. 2007;380:122–127. doi: 10.1016/j.cca.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 13.Sharma G, Burke J, Bhattacharyya S, Sharma N, Katyal S, Park RL, Tobacman J. Reduced arylsulfatase B activity in leukocytes from cystic fibrosis patients. Pediatr Pulmonol. 2013;48:236–244. doi: 10.1002/ppul.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharyya S, Kotlo K, Danziger R, Tobacman JK. Arylsulfatase B regulates interaction of chondroitin-4-sulfate and kininogen in renal epithelial cells. Biochim Biophys Acta. 2010;1802:472–477. doi: 10.1016/j.bbadis.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 15.Kotlo K, Bhattacharyya S, Yang B, Feferman L, Tejaskumar S, Linhardt R, Danziger R, Tobacman JK. Impact of salt exposure on N-acetylgalactosamine-4-sulfatase (Arylsulfatase B) activity, glycosaminoglycans, kininogen, and bradykinin. Glycoconjugate J. 2013 doi: 10.1007/s10719-013-9468-8. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharyya S, Tobacman JK. Hypoxia reduces arylsulfatase B activity and silencing arylsulfatase B replicates and mediates the effects of hypoxia. PLoS One. 2012;7:e33250. doi: 10.1371/journal.pone.0033250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwaki J, Minamisawa T, Tateno H, Kominami J, Suzuki K, Nishi N, Nakamura T, Hirabayashi J. Desulfated galactosaminoglycans are potential ligands for galectins: evidence from frontal affinity chromatography. Biochem Biophys Res Commun. 2008;373:206–212. doi: 10.1016/j.bbrc.2008.05.190. [DOI] [PubMed] [Google Scholar]
- 18.Song S, Byrd JC, Mazurek N, Liu K, Koo JS, Bresalier RS. Galectin-3 modulates MUC2 mucin expression in human colon cancer cells at the level of transcription via AP-1 activation. Gastroenterology. 2005;129:1581–1591. doi: 10.1053/j.gastro.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Nangia-Makker P, Tait L, Balan V, Hogan V, Pienta KJ, Raz A. Regulation of prostate cancer progression by galectin-3. Am J Pathol. 2009;174:1515–1523. doi: 10.2353/ajpath.2009.080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hann A, Gruner A, Chen Y, Gress TM, Buchholz M. Comprehensive analysis of cellular gaectin-3 reveals no consistent oncogenic function in pancreatic cancer cells. PLoS One. 2011;6:e20859. doi: 10.1371/journal.pone.0020859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricciardelli C, Mayne K, Sykes PJ, Wyaymond WA, McCaul K, Marshall VR, Tilley WD, Skinner JM, Horsfall DJ. Elevated stromal chondroitin sulfate glycosaminoglycan predicts progression in early-stage prostate cancer. Clin Cancer Res. 1997;3:983–992. [PubMed] [Google Scholar]
- 22.Ricciardelli C, Mayne K, Sykes PJ, Raymond WA, McCaul K, Marshall VR, Horsfall DJ. Elevated levels of versican but not decorin predict disease progression in early-stage prostate cancer. Clin Cancer Res. 1998;4:963–971. [PubMed] [Google Scholar]
- 23.Ricciardelli C, Sakko AJ, Ween MP, Russell DL, Horsfall DJ. The biological role and regulation of versican levels in cancer. Cancer Metastasis Rev. 2009;28:233–245. doi: 10.1007/s10555-009-9182-y. [DOI] [PubMed] [Google Scholar]
- 24.Sakko AJ, Ricciardelli C, Mayne K, Suwiwat S, LeBaron RG, Marshall VR, Tilley WD, Horsfall DJ. Modulation of prostate cancer cell attachment to matrix by versican. Cancer Res. 2003;63:4786–4791. [PubMed] [Google Scholar]
- 25.Kischel P, Waltregny D, Dumont B, Turtoi A, Greffe Y, Kirsch S, De Pauw E, Castronovo V. Versican overexpression in human breast cancer lesions: known and new isoforms for stromal tumor targeting. Int J Cancer. 2010;126:640–650. doi: 10.1002/ijc.24812. [DOI] [PubMed] [Google Scholar]
- 26.Domenzain-Reyna C, Hernández D, Miquel-Serra L, Docampo MJ, Badenas C, Fabra A, Bassols A. Structure and regulation of the versican promoter: the versican promoter is regulated by AP-1 and TCF transcription factors in invasive human melanoma cells. J Biol Chem. 2009;284:12306–12317. doi: 10.1074/jbc.M807108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holzberg D, Knight CG, Dittrich-Breiholz O, Schneider H, Dörrie A, Hoffmann E, Resch K, Kracht M. Disruption of the c-JUN-JNK Complex by a cell-permeable peptide containing the c-JUN δ domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes. J Biol Chem. 2003;278:40213–40223. doi: 10.1074/jbc.M304058200. [DOI] [PubMed] [Google Scholar]
- 28.Fanjul A, Dawson MI, Hobbs PD, Jong L, Cameron JF, Harley E, Graupner G, Lu XP, Pfahl M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature. 1994;372:107–111. doi: 10.1038/372107a0. [DOI] [PubMed] [Google Scholar]
- 29.Du WW, Yang BB, Shatseva TA, Yang BL, Deng Z, Shan SW, Lee DY, Seth A, Yee AJ. Versican G3 promotes mouse mammary tumor cell growth, migration, and metastasis by influencing EGF receptor signaling. PLoS One. 2010;5:e13828. doi: 10.1371/journal.pone.0013828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y, Chen L, Cao L, Sheng W, Yang BB. Overexpression of the C-terminal PGM/versican domain impairs growth of tumor cells by intervening in the interaction between epidermal growth factor receptor and beta1-integrin. J Cell Sci. 2004;117:2227–2237. doi: 10.1242/jcs.01057. [DOI] [PubMed] [Google Scholar]
- 31.Xiang YY, Dong H, Wan Y, Li J, Yee A, Yang BB, Lu WY. Versican G3 domain regulates neurite growth and synaptic transmission of hippocampal neurons by activation of epidermal growth factor receptor. J Biol Chem. 2006;281:19358–19368. doi: 10.1074/jbc.M512980200. [DOI] [PubMed] [Google Scholar]
- 32.Roeser D, Preusser-Kunze A, Schmidt B, Gasow K, Wittmann JG, Dierks T, von Figura K, Rudolph MG. A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme. Proc Natl Acad Sci US A. 2006;103:81–86. doi: 10.1073/pnas.0507592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roeser D, Schmidt B, Preusser-Kunze A, Rudolph MG. Probing the oxygen-binding site of the human formylglycine generating enzyme using halide ions. Acta Crystallogr D Biol Crystallogr. 2007;63:621–627. doi: 10.1107/S0907444907009961. [DOI] [PubMed] [Google Scholar]
- 34.Wòjczyk B. Lysosomal arylsulfatases A and B from horse blood leukocytes: purification and physico-chemical properties. Biol Cell. 1986;57:147–152. doi: 10.1111/j.1768-322x.1986.tb00471.x. [DOI] [PubMed] [Google Scholar]
- 35.Rao GJ, Christe M. Inhibition of rabbit liver arylsulfatase B by phosphate esters. Biochim Biophys Acta 1994. 1994;788:58–61. doi: 10.1016/0167-4838(84)90297-8. [DOI] [PubMed] [Google Scholar]
- 36.Christianson TM, Starr CM, Zankel EC. Overexpression of inactive arylsulphatase mutants and in vitro activation by light-dependent oxidation with vanadate. Biochem J. 2004;283:581–587. doi: 10.1042/BJ20040447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoo BC, Hong SH, Ku JL, Kim YH, Shin YK, Jang SG, Kim IJ, Jeong SY, Park JG. Galectin-3 stabilizes heterogeneous nuclear ribonucleoprotein Q to maintain proliferation of human colon cancer cells. Cell Mol Life Sci. 2009;66:350–364. doi: 10.1007/s00018-009-8562-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Inohara H, Pienta KJ, Raz A. Galectin-3 is a nuclear matrix protein which binds RNA. Biochem Biophys Res Commun. 1995;217:292–303. doi: 10.1006/bbrc.1995.2777. [DOI] [PubMed] [Google Scholar]
- 39.Harmatz P, Giugliani R, Schwartz IV, Guffon N, Teles EL, Miranda MC, Wraith JE, Beck M, Arash L, Scarpa M, et al. MPS VI Study Group. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) J Pediatr. 2004;144:574–580. doi: 10.1016/j.jpeds.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 40.http://craniofacial.jax.org/publications/curtainetalnm3775m1J/html
- 41.http://primer3.wi.mit.edu
- 42.Rauscher FJ, 3rd, Sambucetti LC, Curran T, Distel RJ, Spiegelman BM. Common DNA binding site for FOS protein complexes and transcription factor AP-1. Cell. 1998;52:471–480. doi: 10.1016/s0092-8674(88)80039-4. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharyya S, Borthakur A, Dudeja PK, Tobacman JK. Carrageenan induces cell cycle arrest in human intestinal epithelial cells in vitro. J Nutr. 2008;138(3):469–475. doi: 10.1093/jn/138.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]