

Abstract
The ability to design artificial extracellular matrices as cell instructive scaffolds has opened the door to technologies capable of studying cell fate in vitro and to guide tissue repair in vivo. One main component of the design of artificial extracellular matrices is the incorporation of biochemical cues to guide cell phenotype and multicellular organization. The extracellular matrix is composed of a heterogeneous mixture of proteins that present a variety of spatially discrete signals to residing cell populations. In contrast, most engineered ECMs do not mimic this heterogeneity. In recent years the use of photodeprotection has been used to achieve spatial immobilization of signals. However, these approaches have been limited mostly to small peptides. Here we combine photodeprotection with enzymatic reaction to achieve spatially controlled immobilization of active bioactive signals that range from small molecules to large proteins. A peptide substrate for transglutaminase factor XIII (FXIIIa) is caged with a photodeprotectable group, which is then immobilized to the bulk of a cell compatible hydrogel. With the use of focused light the substrate can be deprotected and used to immobilize patterned bioactive signals. This approach offers an innovative strategy to immobilize delicate bioactive signals, such as growth factors, without loss of activity and enables In situ cell manipulation of encapsulated cells.
Keywords: Photopatterning, enzymatic reaction, hydrogels, ortho-nitrobenzyl, Factor XIIIa
Introduction
Advances in developmental, cell and molecular biology are beginning to map out the spatial and temporal regulation of bioactive and physical signals required for the orchestration of tissue formation.[1] This spatial regulation of bioactive signals is most dramatic during embryogenesis where a whole organism is created from just one cell. However, the fine tuned orchestration of tissue formation occurs in adults as well during homeostasis and to allow wound healing. It is clear from this wealth of literature that one signal and one static environment are not sufficient for tissue formation or homeostasis. Thus, our ability to probe human cell fate in vitro and determine the minimum required signals for tissue formation or to promote a desired cell phenotype is limited by our inability to create cellular microenvironments with complex and dynamic patterns of signals.
Engineered extracellular matrixes (eECMs) aim to bridge the gap between in vivo studies with those performed in tissue culture plastic. These engineered extracellular matrices (eECMs) can be developed through the use of synthetic polymers or modification of existing natural polymers.[2] Studies in eECMs have opened the door for the understanding of how changes in the bulk physical and chemical characteristics of these three-dimensional (3D) environments influence tissue formation,[3] stem cell differentiation,[2a]pathological conditions,[2b, 2c] and drug delivery.[4] Although eECMs have enable us to study cell behaviour in an environment closer to what they experience in vivo, most still fail to recapitulate the heterogenous nature of the natural ECM both in the spatial control of signal placement and temporal control of signal availability.
To introduce heterogeneity several approaches have been explored using light-controlled platforms to modify the physical and chemical properties of hydrogels. Light-based platforms can allow for modifications ranging from the bulk to micrometer-sized resolution (<10 μm) due to the high level of control available for the stimulus. These platforms fall into two basic modes: subtraction or addition. Photolytic “subtraction” strategies depend on the breaking of a covalent bond to either decrease hydrogel crosslinking density (physical) or release an attached signal (chemical).[5] The Anseth group used this strategy to control the growth of encapsulated mesenchymal stem cells.[5b] Subtractive methods are difficult to use for generating precise spatially-limited chemical patterns, primarily due to the large quantity of deprotection needed to eliminate unwanted signal. In contrast, the “addition” mode only presents the desired signal in the areas exposed to light. Previous, additive methods have relied on either activation of photopolymerizable groups (e.g. acrylates) or uncaging of protected thiols for controlled Michael-type reactions. One of the more successful techniques, employed by the Shoichet group, has allowed for the attachment of multiple proteins within hydrogel matrices.[5d] Beyond this exception, the vast majority of additive strategies have focused on the addition of small oligopeptides. The absence of complex signal patterning is not surprising, as the use of more complex signals (e.g. growth factors) is limited by molecule stability during synthetic modification and purification, as well as the possibility of decreased signal activity as a result of modification and immobilization onto the scaffold. A less explored approach to pattern signals into hydrogel scaffolds is the use of protected peptides that are substrates for enzymes, such that in the presence of the protective group no enzymatic activity is observed but upon light exposure the enzyme can recognize the peptide. This approach was first explored using caged peptide substrates for caspase.[6] Upon photodeprotection and enzymatic digestion of the light exposed areas, the free amine resulting from the digestion was used to immobilize RGD or biotin. Although this report only demonstrated two-dimensional (2D) patterning and used the technology for cell sorting rather than tissue regeneration applications, it demonstrated the ability to use caged peptides to control enzymatic activity within hydrogel scaffolds.
Herein we present a novel approach, termed Hybrid Photopatterned Enzymatic Reaction or HyPER, to immobilize bioactive signals with spatial control into cell compatible hydrogel scaffolds. Our platform uses activated transglutaminase factor XIII (FXIIIa) catalysed reaction to immobilize signals to light-activated regions of the hydrogel. To control enzymatic activity we employ a caged enzyme substrate that is immobilized to the backbone of the hydrogel matrix. The cage prevents enzymatic attachment of the signal before light exposure. Following localized removal of the light-sensitive cage, FXIIIa catalyses the formation of a stable amide bond between the desired bioactive signal and the hydrogel backbone.
Results and Discussion
FXIIIa chemistry and HyPER
FXIIIa is an important enzyme in the blood coagulation cascade and is partially responsible for clot stability through the introduction of additional covalent bonds.[7] Building from this natural capacity, FXIIIa has previously been used for crosslinking and bulk biomolecule immobilization within hydrogels.[2a, 8] More specifically, FXIIIa catalyzes the formation of a stable non-canonical amide bond between the ε-amine of lysine (K) and the γ-carboxamide of glutamine (Q). For our platform the FXIIIa-associated lysine is contained within an oligopeptide, Ac-FKGGERC-NH2[8d] (or K-peptide), which includes a cysteine (C) to allow for pseudo-Michael addition attachment to any hydrogel backbone polymer modified with a suitable vinyl group (Fig. 1A). The FXIIIa associated glutamine is associated with the bioactive signal to be immobilized and is contained within the peptide sequence NQEQVSPL[8a, 9] (or Q-peptide). Both of these sequences were previously identified to be either a synthetic substrate for transglutaminase (K-peptide) through a rational peptide library[8d] or identified as a natural substrate for FXIIIa and have been previously used to immobilize bioactive signals within hydrogel scaffolds[2a, 8a, 8b, 9] or to form hydrogel scaffolds.[8c, 8d]
Figure 1.

Hybrid Photopatterned Enzymatic Reaction (HyPER) achieves predictable immobilization kinetics. A) HyPER utilizes a caged peptide substrate for FXIIIa (K*-peptide) that prevents FXIIIa activity unless the substrate has been deprotected by 365nm light exposure. This allows for site specific deprotection of the K*-peptide. Following deprotection bioactive signal immobilization can be achieved by adding the second substrate for FXIIIa to the bioactive signal of interest and incubating the K-peptide modified hydrogel, the Q-peptide bioactive signal and FXIIIa. B) Synthesis of the photocaged group and direct peptide modification. C) K*-peptide photodeprotection kinetics. D) FITC-Q-RGD was immobilized to hydrogels with either 100% or 50% deprotection of K* using FXIIIa. The amount bound for the 50% was half of that for 100% deprotection (matching the predicted value, shown in black), indicating that the photo deprotection and bioactive signal conjugation can be predicted.
Caged K-peptide substrate synthesis
To allow for photopatterning we caged the ε-amine of lysine with a photoactive ortho-nitrobenzyl (o-NB) vanillin derivative, (4-(4-formyl-2-methoxy-5-nitrophenoxy)butanoic acid).[11] The o-NB cage prevents the FXIIIa catalyzed attachment of the glutaminyl peptide residue in the Q-peptide. The o-NB cage was chosen for its high absorption spectra (λpeak~350nm) and fast degradation time (t1/2 ~7–8m at 10 mW/cm2),[11–12] which minimizes the risk of damage to present biomolecules (e.g. DNA). In addition, o-NBs are susceptible to multiphoton degradation, allowing for true 3D patterning within hydrogels.[5b, 5c] Once the cage is removed via photo-deprotection, the lysine returns to a primary amine capable of transglutaminase modification (Fig. 1A).
The o-NB molecule was synthesized from vanillin. Briefly, we etherify vanillin via a Williamson etherification with ethyl-4-bromobutyrate. This is followed by nitration at ortho-position with nitric acid to provide light sensitivity. Next the nitrated product is hydrolyzed via trifluoroacetic acid to produce a more water soluble material before reacting with the full length peptide (Fig. 1B). We caged the ε-amine residue of the K-peptide through reductive amination, using picoline borane to directly modify the full length peptide in a single reaction that results in a high yield (40–50%) following preparatory HPLC purification of the caged peptide (Fig. 1B). The absence of unmodified K-peptide along with the overall purity of the product was verified by electrospray ionization mass spectrometry and LCMS (see Supplemental).
Caged K-peptide photo-deprotection kinetics and FXIIIa immobilization can be predicted
To determine the extent to which we could predict the amount of signal that would be immobilized following photo-deprotection and FXIIIa mediated immobilization, we first explored the light-controlled uncaging kinetics of the protected K-peptide using a soluble model system. Caged K peptide was exposed to 365 nm filtered UV light (I = 20 mW/cm2). At different exposure times, samples (n = 3) were analyzed for the presence of free amines on the uncaged lysine residues via reaction with trinitrobenzene sulfonate (TNBS), a common assay for primary amine quantification[13] (Fig. 1C). Using a simplified equation for exponential degradation[14] (equation 1) we determined the intensity dependent degradation constant of the caged K-peptide (k = 0.00024 sec−1(mW/cm2)−1), which is comparable to previously published results of o-NB groups derived from vanillin (k = 0.00015; k = 0.00026).[11–12] At 20 mW/cm2, this degradation constant results in a t1/2 of ~208 s, which can be increased by lowering the light intensity (e.g. 10 mW/cm2: t1/2 = 416 s) or decreased by increasing the light intensity.
| (Eq. 1) |
We wanted to next determine if with a defined degradation constant we could predict the amount of bioactive signal immobilized via FXIIIa in a cell compatible hydrogel. To determine the predictability of the system, we ran an immobilization test within a hyaluronic acid-based (HA) hydrogel (1.6 mM caged K peptide, K*) using a fluorescently tagged Q-RGD substrate and measuring depletion of surrounding fluorescence to determine immobilization (Fig. 1D). This HA hydrogel has been extensively used in our laboratory[3a, 4, 15] for the culture of stem cells and is synthesized through Michael type addition of acrylates present in the HA backbone and dithiol containing peptides. One set of hydrogels was fully uncaged (I = 20 mW/cm2, t = 1200 s), while a second was exposed to enough light to theoretically achieve 50% uncaging (I = 10 mW/cm2, t = 416 s). Both samples were exposed to FXIIIa and FITC-Q-RGD for conjugation. The fully uncaged gels displayed 0.446±0.085 mM Q-peptide immobilized, thus we expect the 50% uncaged set to display ~0.223±0.042 mM Q-peptide, which is what we observed (0.253±0.024 mM) (Fig 1D). However, this also demonstrated that even with full deprotection we could not achieve 100% utilization of the deprotected K-peptides via FXIIIa chemistry with an efficiency of only 27.2%. As controls, the same hydrogels were used with either no light exposure (0% uncaged) or no K-peptide added but with FXIIIa. The unexposed hydrogel showed no statistically significant attachment of Q-peptide over the control without K-peptide and both controls were statistically lower than those exposed to light and containing both FXIIIa and K-peptide. Thus, the predictable kinetics of deprotection and the FXIIIa catalyzed reactions allow for predictable immobilization.
HyPER can achieve spatial co-immobilization of multiple bioactive signals under cell compatible conditions
We next wanted to determine if we could use HyPER to immobilize bioactive signals ranging in molecular weight and complexity using either filtered 365 nm light or a two-photon confocal microscope. For these experiments we used either four-arm PEG-vinyl sulfone (PEG-VS) or HA-acrylate (HA-AC) both crosslinked through Michael addition using dithiol containing peptides. Our chosen substrates included an RGD adhesive peptide (Q-RGD), a fibronectin protein fragment (Q-FNIII9*-10),[2b, 2c, 10] and two growth factors (vascular endothelial growth factor, Q-VEGF, and platelet-derived growth factor, Q-PDGF). The Q-peptide sequence was introduced either during solid phase peptide synthesis for smaller peptides containing a bioactive signal, or by introducing the sequence at the N-terminus of larger proteins prepared using standard recombinant protein cloning and expression techniques[8b] (supplementary information).
For all the immobilization experiments, 1.6 mM K*-peptide was immobilized to the hydrogel backbone and FXIIIa enzyme was added to the hydrogel prior to gelation. The hydrogel was formed using 4 % HA and an r-ratio of 0.6, which resulted in an elastic modulus of 300 Pa. We first used a mercury lamp at 10 mW/cm2 intensity in combination with a photomask to deprotect the desired 2D areas within the hydrogel (Fig. 2A). Following deprotection the hydrogel was exposed to the conjugation solution that contained the bioactive signal to be immobilized. After conjugation, the hydrogels were washed to remove un-bound material. We first use PEG-VS to immobilize the small molecule NHS-Alexa 556. This immobilization does not use the HyPER platform, rather it uses the deprotected amines for conjugation (Fig2B). This data shows that deprotection is possible an efficient and leads to the generation of a free amine. Next we used HA-AC/MMP hydrogels to immobilize through FXIIIa. Using HyPER were able to immobilize fluorescently-labelled Q-RGD in circles and a dumbbell, (Fig. 2 C,D) and Q-FNIII9*-10 in lines (Fig. 2E) with 2D control over patterning.
Figure 2.

Immobilization of bioactive signals with different molecular weights using HyPER. A) Schematic of HyPER bioactive signal immobilization in hydrogel scaffolds using a photomask. B–D) Immobilization of Q-RGD (MW) in different patterns. E) Immobilization of an engineered fibronectin fragment FN9*10 (MW).[2b, 2c, 10] F) dual pattern of Q-RGD (purple) and Q-PDGF (green). G) linear gradient of Q-RGD. H) Quantification of the linear gradient of Q-RGD. I) Schematic of two-photon deprotection of K*-modified hydrogels. J) Two-photon patterning of RGD. Size bar for all images is 200μm.
We next wanted to determine if HyPER could achieve sequential immobilization of signals and immobilization in a gradient. We use the approach described above, except that after photo-deprotection and immobilizing Q-RGD in lines and washing the hydrogel a second deprotection with 90° lines was performed followed by a second incubation with fluorescently-labelled Q-PDGF. We were able to immobilize both bioactive signals (Fig. 2F), demonstrating that the FXIIIa enzyme remains active post the first modification and allowed a second conjugation to take place. Dual factor immobilization is essential for the generation of complex eECM environments since many bioactive signals are synergistic when placed in close proximity. In order to generate a continuous gradient we used a neutral density continuous gradient optics filter to pattern the hydrogel and immobilize Q-RGD. We show a continuous gradient of Q-RGD, (Fig. 2G). Converting the image to gray scale and determining pixel intensity as a function of distance performed the quantification of the gradient (Fig 2H). Gradients are of great importance in biological systems since they have been shown to be major morphogenic signals during development,[16] adult healing,[17] and disease.[18]
In addition to 2D patterns, we wanted to test the ability of our K* peptide to undergo multiphoton uncaging. Two-photon confocal has been previously shown to be able to deprotect o-NB groups, resulting in 3D controlled patterning.[5c, 5d] Using a similar strategy as detailed above, except that uncaging is achieved using a two photon confocal microscope at 730 nm (twice λpeak). Patterning was achieved by setting a region of interest (ROI) and performing multiple scans (~40 scans, Fig. 2I). Q-VEGF was immobilized in true 3D patterning (Fig. 2J) as shown by the depth dependence of the pattern focus area. The VEGF pattern was 180 μm wide and 75 μm high, with lettering as thin as 6 μm and an overall z-axis depth of ~20 μm (Fig 2J).
The sharpness of the patterns can be improved with enhancing the accessibility of the K-peptide
Although we were able to immobilize all the bioactive signals with spatial control using HyPER, the pattern sometimes was not as sharp as we expected. As we quantified above, our current method only has 27.2% efficiency. We hypothesized that the reason for the low attachment was the access of the K-peptide substrate to the enzyme. To test this hypothesis we changed the presentation of the K*-peptide to have a longer more flexible arm from the HA backbone by modifying fewer of the molecules of HA (6.8% versus 100%) with the K* peptides (~50 K*-peptide per modified HA molecule versus ~3.5 K* peptide per modified HA molecule). The visualization of FXIIIa immobilized Q-RGD and Q-VEGF clearly demonstrates the increased binding when the K*-peptide is immobilized in a clustered conformation, rather than homogeneous showing a much sharper pattern with less background (Fig 3).
Figure 3.

Clustering of K*-peptide enhances attachment of bioactive signals through HyPER. Cartoons show the concept of homogeneous and clustered K*-peptide immobilization. Top row, homogeneous K*-peptide immobilization, shows less attachment (less defined patterns and higher background) than the bottom row that show the attachment for when K*-peptide is clustered.
Using HyPER for in situ cell manipulation
We next wanted to determine if the bioactive signals immobilized are active. We first tested if RGD modified gels resulted in cell attachment of mouse mesenchymal stem cells to HyPER modified hydrogels (mMSCs, Fig. 4A–F). K*-peptide modified gels were formed and were uniformly exposed to light (Fig 4A,B) or not exposed to light (Fig 4.C,D). For both types of gels we incubated the gels with the Q-RGD conjugation solution or the Q-RGD conjugation solution minus FXIIIa. Following conjugation mMSCs were seeded on the surface of the gels and phase images were taken at day 3. Only those cells that were cultured on fully exposed hydrogels with complete conjugation solution including FXIIIa resulted in significant spreading similar to our positive control that contains RGD immobilized prior to hydrogel gelation as traditionally done in our lab.[15] These results also illustrate the specificity of the FXIIIa reaction with those gels that had light exposure but no FXIIIa resulting in no spreading (Fig 4A). To determine if the cells could respond to a pattern or RGD, 100 μm circles of Q-RGD were patterned (Fig 4. G–I). mMSCs cultured on the surface of these gels showed self-organization of the cells into the patterned regions (Fig. 4I).
Figure 4.

Bioactive signals immobilized through HyPER retain their activity. A–F) mMSCs are seeded on the surface of Q-RGD modified surfaces using the conditions indicated underneath each image. B) Only when light exposure is combined with Q-RGD and FXIIIa do cells spread as well as when E) RGD is introduced to the bulk of the hydrogel. G) RGD patterned in 100μm circles, H) cells cluster to the location of the pattern, I) overlay of the pattern and cell cluster. J–M) HUVECs are cultured on the surface of Q-VEGF modified hydrogels. Hydrogels were treated with decreasing amounts of light to result in 80% to 0% deprotection and QVEGF was immobilized using HyPER. J) 80% and K) 40% deprotection resulted in more cells at the cell surface than L) 10% and M) 0% deprotection. O) HUVEC proliferation was quantified. Statistical analysis was performed using an ANOVA with a Tukey post-test. * and ** indicate that that condition is significantly different from every other condition by at least p < 0.05 or 0.01, respectively.
To investigate the bioactivity of immobilized Q-VEGF, increasing amounts of Q-VEGF were immobilized to K*-peptide functionalized hydrogels that were deprotected to achieve 0, 10, 40 and 80% deprotection. We expected that human umbilical vein endothelial cells (HUVECs) seeded on the VEGF modified hydrogels will proliferate at different rates depending on the quantity of immobilized VEGF. Images taken at day 5, show that both 40% and 80% exposed samples (Fig 4 J,K) contain more cells than the 10% and 0% samples (Fig 4 L, M), supporting our expectations. Cell proliferation was quantified and showed similar proliferation rates for the 80% and 40% deprotected samples, but a statistically higher proliferation compared to 0 and 10% deprotected samples (Fig. 4O), indicating that different amounts of VEGF can be immobilized on the surface and that lower Q-VEGF immobilization results in lower proliferation. Interestingly our negative control that has no light exposure (0% deprotected K*) and Q-VEGF conjugation solution minus FXIIIa resulted in more cell proliferation than the same surfaces but not exposed to Q-VEGF. This result indicates that Q-VEGF has some affinity for hyaluronic acid that results in some background VEGF binding.
Last we wanted to explore the ability to pattern bioactive signals in situ in the presence of cells. For this experiments HA-RGD-K*-peptide hydrogels were synthesized with mMSCs and FXIII plated throughout the hydrogel volume. To determine if HyPER was cell biocompatible, we performed a Q-RGD attachment and performed a LIVE/DEAD stain 1 day post HyPER (Fig 5A–C). Cells that were exposed to light and were subjected to the conjugation solution (Thrombin, Q-RGD, Ca2+), had the same level of LIVE cells as those that were not exposed to light but still had the conjugation solution, which had the same LIVE cells as those that did not have exposure to light or conjugation solution. This results show that neither the exposure to light nor the conjugation solution affect cell viability. We next wanted to look for a cellular response as a function of immobilized Q-PDGF. mMSCs have PDGF receptors and have been shown to respond to PDGF.[19] The RGD peptide was bound using Michael addition and the cells were allowed to spread for one day before the K* was deprotected using fully exposed (no pattern) light or a optical filter gradient (365nm light at 10mW/cm2). Q-PDGF was then immobilized using FXIIIa chemistry using the same approach used in Figure 2 and the cells further cultured for another 2 days before imaging. The cells cultured with the immobilized Q-PDGF gradient resulted in different morphology and actin cytoskeletal staining than those cells cultured in homogenously bound or in the presence of soluble Q-PDGF (Fig 4D–I). Comparing mMSCs cultured in homogenous immobilized Q-PDGF (Fig 4D, G) and cultured in the presence of soluble Q-PDGF (Fig 4F, I) to mMSCs cultured in PDGF immobilized in a gradient (Fig 4E, H), we observe more polarized and extended cells in the cells cultured in the gradient than those cultured in the homogeneous hydrogels or those exposed to soluble PDGF. This experiment highlights the types of experiments that could be performed with HyPER. One could plate individual cells, cell clusters, organoids, etc in the gel, then after a planned period of time a desired signal with the desired pattern can be immobilized to study the effect of that signal on morphogenesis or stem cell differentiation. This type of cell biocompatible and protein compatible patterning is not currently available.
Figure 5.
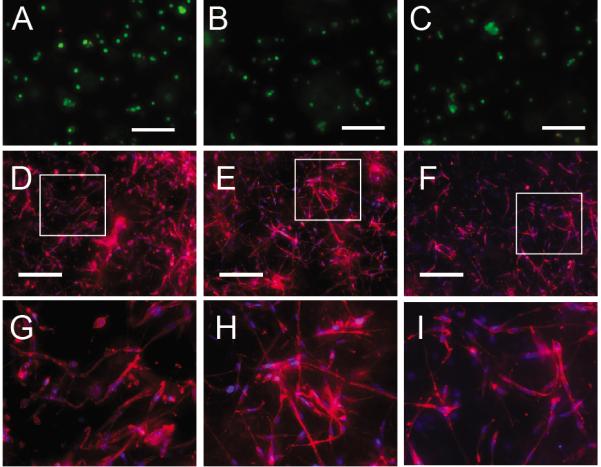
In situ cell manipulation using HyPER. (A–C) Live/Dead Analysis of mMSCs within PEGVS gels with 1.6 mM caged K peptide. Cells showed high viability whether they were exposed to 365 nm light A) with or B) without application of Q-RGD, as did C) the sample left unexposed to either. D–I) In situ hydrogel patterning of Q-PDGF in the presence of cells was performed by first making HA-RGD hydrogels with mMSCs plated homogeneously and 24-hrs later immobilizing Q-PDGF D,G) homogeneously in the hydrogel through complete photo-deprotection or E,H) in a gradient by deprotecting using a radial gradient. F,I) For comparison, mMSCs seeded inside HA-RGD gels were exposed to soluble Q-PDGF. Images G–I) represent the boxed region in D–F).
Conclusion
In summation, we have developed a high yield method for direct caging of a FXIIIa-recognized substrate, K-peptide. Using light we were able to deprotect the FXIIIa substrate and then use FXIIIa to catalyze the immobilization of bioactive signals. Our studies have shown the resultant platform displays quantifiable uncaging behavior, resulting in predictable immobilization behavior. The enzymatic immobilization chemistry is highly modular, allowing for spatially-defined patterns of a wide variety of active signals, including oligopeptides, protein fragments, and growth factors. We demonstrated patterning in 2D using photomasks and optical filters to produce both binary and gradient patterns, respectively, and in 3D via a previously developed multiphoton uncaging technique. Finally, we demonstrated the capacity of our patterning platform to control cell behavior through the alteration of cell morphology, viability, and proliferation. In addition, we demonstrate the ability to modify the microenvironment of cells in situ without cell damage. We believe that the HyPER platform will provide an invaluable tool for future studies of both cell biology and tissue engineering.
Experimental Section
Materials
Hyaluronic acid (60,000 Da, Genzyme Corporation, Cambridge, MA). Adipic dihydrazide, NHS-acrylate, acryloyl chloride, triethylamine, N-boc-ethylenediamine, 4-(4,6-dimethoxy[1,3,5]triazin-2-yl)-4-methylmorpholinium chloride (DMTMM), vanillin, potassium carbonate, ethyl-4-bromobutyrate, dimethylformamide (DMF), nitric acid, ethanol, sulfuric acid, trifluoroacetic acid, methanol, and acetic acid were purchased from Fisher Scientific and used without purification. Picoline borane complex and PEG dithiol (MW 1000 Da) were purchased from Sigma Aldrich and used without purification. Ac-FKGGGERC-NH2 (K peptide), Ac-GCRDGPQGIWGQDRCG-NH2, NQEQVSPLRGDSPGNH2 (Q-RGD peptide), and FITC-labeled NQEQVSPLRGDSPG-NH2 were purchased from Genscript (Piscataway, NJ) and used without purification. 4-arm PEG-vinylsulfone (PEG-VS, MW 40kDa) and 4-arm PEG-maleimide (PEG-MAL, MW 20kDa) were purchased from JenKem Inc. and used without further purification.
Cell culture
Mouse mesenchymal stem cells (mMSCs, ATCC, Cat. CRL-12424) were used between P3 and P6. Human Umbilical Vein Endothelial Cells (HUVECs, Lonza, Cat. CC-2519) were used between P3 and P5. Both cell lines were maintained using manufacturer's protocols.
Ortho-nitrobenzyl intermediate synthesis
Ethyl 4-(4-formyl-2-methoxyphenoxy)butanoate
Vanillin (20.0 g, 131 mmol), potassium carbonate (36.3 g, 263 mmol), and ethyl-4-bromobutyrate (25.2 g, 129 mmol) was dissolved in DMF (100 mL) and stirred overnight. The solution was precipitated into water (2000 mL) and stirred for 2 hours. The resultant precipitate was filtered and washed with water to collect (32.6 g, 123 mmol, 95%) as a white powder. 1H NMR (CDCl3, ppm): δ=9.89 (s, 1H), 7.45 (d, 1H), 7.42 (s, 1H), 6.99 (d, 1H), 4.21 (t, 2H), 4.19 (q, 2H), 3.96 (s, 3H), 2.58 (t, 2H), 2.18 (m, 2H), 1.24 (t, 3H).
Ethyl 4-(4-formyl-2-methoxy-5-nitrophenoxy)butanoate
70% Nitric acid (40 mL) was cooled to 0°C in an ice bath and ethyl 4-(4-acetyl-2-methoxyphenoxy)butanoate (4.0 g, 15.0 mmol) was added slowly for five minutes. The ice bath was then removed and the solution was allowed to warm to room temperature and react for 3 hours. The solution was then precipitated into water and filtered as a yellow precipitate. To purify the product the precipitate was esterified with refluxing ethanol (100 mL) and sulfuric acid (cat.) until TLC (9:1 DCM/EtOAc) indicated product was completely esterified. The solution was then allowed to cool slowly and recrystallize. The resulting crystals were filtered as pale, yellow crystals (3.50 g, 11.3 mmol, 75%). 1H NMR (CDCl3, ppm): δ=10.50 (s, 1H), 7.67 (s, 1H), 7.45 (s, 1H), 4.25 (t, 2H), 4.20 (q, 2H), 4.03 (s, 3H), 2.58 (t, 2H), 2.18 (m, 2H), 1.24 (t, 3H).
4-(4-formyl-2-methoxy-5-nitrophenoxy)butanoic acid
Ethyl 4-(4-formyl-2-methoxy-5-nitrophenoxy)butanoate (5.75 g, 18.4 mmol) was heated to 90°C in a solution of trifuoroacetic acid (5 mL) and water (50 mL) for 3 hours. Once TLC (9:1 DCM/EtOAc) showed complete hydrolysis the solution was allowed to cool slowly, resulting in formation of crystals that did not require further purification. The crystals were filtered as a yellow solid (4.49 g, 15.7 mmol, 86%). 1H NMR (d-DMSO) (Figure S2): δ=12.13 (s, 1H), 10.14 (s, 1H), 7.65 (s, 1H), 7.29 (s, 1H), 4.20 (t, 2H), 3.94 (s, 3H), 2.39 (t, 2H), 1.98 (m, 2H).
Caged K-peptide synthesis
Photocaging of K peptide
Ac-FKGGGERC-NH2 (30 mg, 33.6 μmol), 4-(4-formyl-2-methoxy-5-nitrophenoxy)butanoic acid (28.5 mg, 101 μmol), and picoline borane complex (8.2 mg, 36.9 μmol) are dissolved in of a methanol/acetic acid mixture (1.50 mL, 9:1 volume ratio). The reaction proceeds for 18 hours at room temperature. The product is isolated by preparatory HPLC using a 30 minute 5–95% acetonitrile:water (0.1% TFA) soluble phase gradient (elution time: 15–17 minutes). The product is then lyophilized and evaluated by Ellman's reagent (Thermo Scientific Pierce) to determine product yield and tested by electrospray mass spectrometry (Waters LCT Premier XE time of flight instrument controlled by MassLynx 4.1 software) to determine purity (see Figure S3). The K star peptide was collected as a pale yellow powder (13.0 μmol, 48% yield).
Synthesis of Acrylated Hyaluronic Acid (HA-AC)
N-acryloyl ethylene diamine
In a rb flask with stir bar and i/a Ar add acryloyl chloride (3.03 mL, 37.4 mmol) and chloroform (74.8 mL). Cool with ice-water bath. Add following dropwise: N-boc-ethylene diamine (5.00 g, 31.2 mmol), triethylamine (4.57 mL, 32.8 mmol), and chloroform (37.4 mL). Allow to warm to RT for 2 h. Remove chloroform by rotary evaporation. Add water (50 mL) and mix thoroughly. Extract with chloroform (3×30mL). Dry with magnesium sulfate, filter, and remove chloroform by rotary evaporation. Dissolve in concentrated hydrochloric acid (HCl) (7 mL, ~2 mL/g). Allow to react for 1 h. Place under vacuum overnight to evaporate HCl. Add ethanol (250 mL) and stir for 1 h. Filter and remove ethanol by rotary evaporation. Yield: 1.78 g (11.83 mmol, 38%). 1H NMR (d-DMSO): δ=6.20 (m, 1H), 6.08 (d, 1H), 5.55 (d, 1H), 3.35 (m, 2H), 2.85 (m, 2H).
HA-AC
Following a modified literature procedure:[20] Dissolve HA (2.2 g, 5.8 mmol) in 100 mM MES (pH 5.5). Add DMTMM (3.42 g, 11.6 mmol) and N-acryloyl ethylene diamine (1.75 g, 11.6 mmol). React overnight at RT and dialyze with water for 24 h (4×4 L). Lyophilize to remove water. Yield: 2.1 g (90% yield; 64.45% acrylate modification). Characterize AC modification by 1H-NMR. Determine modification by summing the integration of the three acrylate peaks (1H NMR (d-D2O): δ=6.20 (m, 1H), 6.08 (d, 1H), 5.55 (d, 1H)) and dividing by the integration of the HA acetyl peak (δ=1.85–1.95 (s, 3H).
Degradation kinetics experiment
K star photodegradation kinetics
A 0.2 mM stock solution of the K star peptide was made using PBS. For each time of exposure data point (N = 3), we placed an aliquot of this solution (0.7 mL) between two glass slides separated by 0.25 mm spacers via pipette. The solution was then exposed to 20 mW/cm2 UV light (Irgacure Series 1000, 365 nm internal bandpass filter) for a predetermined period of time. Following exposure, the solution was testing by trinitrobenzosulfonic acid (TNBSA) following the company published protocol (Pierce Thermo Scientific). The results were analyzed using Prism software (GraphPad Software, Inc.) to determine the exponential constant, k, that is in Equation 1.
Hydrogel synthesis techniques
Hydrogel type A (non-clustered) synthesis
Lyophilized acrylated hyaluronic acid (HA-AC Method 2) (64.45% Acrylate modification) was dissolved (0.08 mg/μL) in 0.3 M triethanolamine (TEOA, Fisher Scientific) by vortexing. K star peptide (0.1 μmol) was dissolved in 0.08 mg/μL HA-AC (25 μL) and allowed to react for 20 min at 37°C. To this solution was added TEOA (37.1 μL) and Factor XIIIa (2.78 μL, 180 U/mL). This solution was then added to an aliquot of MMP crosslinker (1.07mg, Ac-GCRDGPQGIWGQDRCG-NH2), vortexed, aliquoted (10 μL) between two Sigmacote (Sigma–Aldrich) surface functionalized glass coverslips and placed in an incubator for 45 min at 37°C to gel (Note: For best results, following mixing of all gel components solution pH should be between 7.9 and 8.2). Following gelation the hydrogels were equilibrated in PBS w/ CaCl2 (0.9 mM).
Hydrogel type B (clustered) synthesis
Lyophilized acrylated hyaluronic acid (HA-AC Method 2) (64.45% Acrylate modification) was dissolved (0.08 mg/μL) in 0.3 M triethanolamine (TEOA, pH 9.2, Fisher Scientific) by vortexing. K star peptide (0.1 μmol) was dissolved in 0.08 mg/μL HA-AC (1.71 μL) and 22.2 μL TEOA and allowed to react for 45 min at 37°C. To this solution was added 23.29 μL HA-AC (0.08 mg/μL) and 2.78 μL Factor XIIIa (180 U/mL). This solution was then added to an aliquot of MMP crosslinker (1.07mg, Ac-GCRDGPQGIWGQDRCG-NH2), vortexed, aliquoted (10 μL) between two Sigmacote (Sigma–Aldrich) surface functionalized glass coverslips and placed in an incubator for 45 min at 37°C to gel (Note: For best results, following mixing of all gel components solution pH should be between 7.9 and 8.2). Following gelation the hydrogels were equilibrated in PBS w/ CaCl2 (0.9 mM).
Hydrogel type C (PEG) synthesis
PEG-MAL and RGD and K star peptide were dissolved in PBS by vortexing to get stock concentrations of 10 mM, 5 mM, and 5 mM, respectively. For a typical gel (15μL, 2 mM (4wt%) PEG-MAL and 1 mM of each oligopeptide) aliquots of each oligopeptide stock (3μL) and of PEG-MAL stock (3μL) were combined and allowed to react for 5 m at 37°C. The remaining PBS volume (4.40μL) was then added to reach the desired final concentrations. A MMP crosslinker (Ac-GCRDGPQGIWGQDRCG-NH2) was dissolved in PBS to a stock concentration of 28.2 mM. The MMP crosslinker stock (1.60 μL) was added according to the desired r-ratio (thiol:maleimide) 0.75 (3 mM MMP crosslinker), vortexed and the 15μL gel was pipetted quickly between Sigmacote (Sigma–Aldrich) surface functionalized glass coverslips separated by 1 mm spacers and placed in an incubator for 10 min at 37°C to gel. The gels were swelled overnight in PBS (0.9 mM CaCl2).
Note
For acrylate-thiol and vinylsulfone-thiol Michael addition chemistry we found the efficiency of the reaction to be approximately 80%. For example, for 2 mM peptide incorporated into the gelling procedure, we expect 1.6 mM attached peptide due to a competing disulfide formation reaction (with the remainder washing out of the gel via diffusion).
Enzymatic immobilization experiment
Enzymatic attachment quantification
K star hydrogels (Hydrogel type A) were synthesized as described above (N = 12). The hydrogels were split into three groups dependant on exposure to 10 mW/cm2 UV light (Omnicure Series 1000, 365 nm internal bandpass filter): fully caged, fully uncaged, and partially uncaged. The fully caged set (negative control) was not exposed. The fully uncaged set (positive control) was exposed for 30 minutes. The partially uncaged set was exposed for 5 minutes. The hydrogels were then exposed to a 1 mM solution of FITC-labeled Q-RGD in Tris-buffered saline with 50 mM CaCl2 and 1 U/mL of Thrombin for 3 hours. The fluorescence of the surrounding solution was measured for all sets. The fractional change in fluorescence for the surrounding solution (compared to the set without K peptide) was used to determine the immobilized fraction.
Predicting immobilization with Eq 1
To predict the expected Q-RGD immobilized shown in Figure 1D, we first determined the theoretical uncaged fraction using Equation 1 with k = 0.24 × 10−4 sec−1 (mW/cm2)−1 (as determined in Figure 1C), I = 10 mW/cm2, and t = 417 seconds. We then multiplied the theoretical uncaged fraction (0.5 for this example) times the concentration of Q-RGD immobilized for the fully deprotected sample 446±85 μM to provide the expected concentration for the partially uncaged sample (0.223±42 μM).
General patterning technique used for Fig. 2
UV Single photon Patterning
Hydrogels (type A for images 2C–F, type C for 2B) were placed directly onto photomask surface. An Omnicure 1000 mercury vapor lamp (EXFO) equipped with an internal 365 nm narrow bandpass filter delivered UV light (10 mW/cm2) for 10 minutes to achieve patterned K star deprotection through the photomask. Light intensity was measured with a UVX digital radiometer (UVP). For creation of immobilized gradients, a reverse bullseye filter (Edmund Optics) was used in place of a photomask.
Confocal Multi-Photon Patterning
We used a modified published protocol to pattern the hydrogels (type A) by multiphoton exposure.[21] Briefly, a Zeiss Confocal CTR MIC microscope was used to create specific designed voids within hydrogels. Regions of interest were drawn within the Confocal Software for scanning using the multiphoton laser using a 40× oil immersion objective (730 nm, Intensity setting = 20, 40 scans).
Alexa fluor 555 staining (Fig. 2B)
Following patterning, hydrogels (Gel type C) were incubated in a 10 μg/mL solution of NHS-Alexa 555 (succinimidyl ester alexa 555, Invitrogen) for 2hours, washed overnight in 1×PBS, and imaged on a Zeiss Confocal CTR MIC microscope.
Peptide and Protein Immobilization
Following patterning, hydrogels (Figure 2 used Gel type A and Figure 3 used Gel type B) were exposed to 10 μL of conjugation solution (4 mM Q-functionalized peptide or 100 μg/mL Q-functionalized protein, PBS, 0.9 mM CaCl2) for 3 hours at 37°C. The hydrogels then underwent successive washes of PBS until background fluorescence was removed.
Q-RGD patterning for controlling mMSC morphology
Hydrogel preparation
Hydrogels were synthesized as described above (Gel type B) with the exception that no FXIIIa was included within the gel (FXIIIa was included with the Q-RGD immobilization solution) and the MMP crosslinker was exchanged for a solution of PEG dithiol (MW 1000, Sigma Aldrich) (7.31 μL, 0.2 mg/μL) with the excess volume being subtracted from extra TEOA. Two sets were left caged, two were uncaged (671 sec or 80% theoretical uncaging), and one set was patterned with 100 μm circles. Only one of each caged and fully uncaged set (and the patterned set) was exposed to a Q-RGD (NQEQVSPLRGDSP, 10% FITC-labeled) conjugation solution following the previously mentioned peptide patterning technique. This resulted in three negative gel sets (+UV/−Q-RGD, −UV/+Q-RGD, and −UV/−Q-RGD) and one positive (+UV/+Q-RGD). In addition a positive control (synthesized following Gel type C protocol with 1.6 mM Ac-GCREGRGDSPG-NH2 instead of 1.6 mM K star peptide) and a negative control (synthesized following Gel type C protocol without addition of K star peptide). The hydrogels were washed overnight with PBS (1 mL per gel) before seeding.
Hydrogel seeding
The hydrogels were seeded with a droplet of 10 μL complete medium (CDMEM, DMEM w/ 10% BGS and 0.1% P/S) with mouse mesenchymal stem cells (mMSCs, 4,000 per gel). The cells were allowed to settle for 1 hour before addition of 0.5 mL complete medium and culturing for 72 hours. The cells were then imaged for analysis using a Zeiss Observer Z1 fluorescent microscope.
Q-VEGF patterning for controlling HUVEC viability and proliferation
Hydrogel preparation
Hydrogels were synthesized as described above (Gel type B) with the addition of 0.2 mM Ac-GCREGRGDSPG-NH2. The following exposure conditions were used (0%, 10%, 40%, and 80% uncaged; N = 6 per condition). Half of the gels (N = 3) of each condition were placed directly into PBS with 0.9 mM CaCl2 to act as a negative control (no Q-VEGF). The rest of the hydrogels underwent Q-VEGF immobilization as described above with the exception that FXIIIa (10 U/mL) was included with the Q-VEGF. The hydrogels were washed with PBS (1 mL per gel) for 48 hours at 5°C, with solution changes every ~12 hours.
Hydrogel seeding
The hydrogels were seeded with a droplet of 10 μL EGM-2 (w/out VEGF) with Human Umbilical Vein Endothelial Cells (HUVECs, 4,000 per gel). The cells were allowed to settle for 1 hour before addition of 0.5 mL EGM (w/out VEGF) and culturing for 5 days. The cells were then imaged at Day 5 using a Zeiss Observer Z1 fluorescent microscope.
Proliferation analysis
Cell proliferation was quantified using the CyQUANT® Cell Proliferation Assay by Life Technologies. The results were analyzed using Prism software (GraphPad Software, Inc.) and a one-way ANOVA test was used to determine significance.
LIVE/DEAD analysis of Q-RGD patterning procedure
Hydrogel preparation
Hydrogels were synthesized with inclusion of mMSCs (1500 cells per μL) using PEG-VS. The PEG-VS stock (20 wt%) was dissolved in HEPES buffer (pH 8.2, 0.3M). A stock solution of caged k peptide (5 mM) and a stock solution of MMP crosslinker (0.05 mg/μL) were dissolved in PBS. For a typical gel (45 μL) PEG-VS (13.5 μL), caged K peptide (9 μL), Factor XIIIa (10 μL, 100 U/mL), and MMP crosslinker (6.95 μL) were combined and aliquoted (10 μL) between glass slides (1 mm spacer). Following a 30 m incubation period at 37 °C, the hydrogels were equilibrated and cultured in CDMEM.
Hydrogel patterning and immobilization
Two sets of hydrogels (N = 3) was exposed for 600 s to 365 nm light (10 mW/cm2) and a third set was left unexposed. One of the exposed sets underwent the peptide immobilization procedure detailed above for Q-RGD. All sets were washed with CDMEM (1 mL per gel) twice while being incubated over the following 24 hours.
LIVE/DEAD analysis
Following 24 hours of incubation, the hydrogels were tested by LIVE/DEAD assay (Life Technologies) following the manufacturer's protocol. Hydrogels were then imaged using a Zeiss Observer Z1 fluorescent microscope.
PDGF immobilization with encapsulated mMSCs
Hydrogel preparation
Hydrogels (N = 9) were synthesized following type A synthesis, except that 30% of the gel volume consisted of CDMEM (this volume was subtracted from the total TEOA volume added) with cells (5000 cells per μL). Following gelation the gels were incubated in CDMEM for 24 h.
Hydrogel patterning
One set of hydrogels (N = 3) was exposed to 10 mW/cm2 of 365 nm light passed through two stacked Apodizing Reverse Bullseye optical filters (Edmund Optics, 0.04 – 2.0 OD, 25 mm) for 600 s to produce a theoretical gradient of ~60–10% uncaged from inside to outside of gel (~4 mm radius). A second set (N = 3) was exposed uniformly to 10 mW/cm2 for 600 s (~75% deprotection), while the third set (N = 3) was left unexposed.
Immobilization and culturing
All three sets were exposed to Q-PDGF (10 μL, 100 μg/mL, PBS, 0.9 mM CaCl2) following the above mentioned protein immobilization procedure with PBS washes replaced with three CDMEM washes over 2 days at which point the hydrogels were fixed with 4% PFA.
Staining and imaging
Fixed hydrogels were stained using manufacturer's protocols for rhodamine phalloidin (Life Technologies) and DAPI (Life Technologies) to label cell actin and nuclei, respectively. Hydrogels were then imaged using a Zeiss Observer Z1 fluorescent microscope.
Supplementary Material
Acknowledgements
We like to thank our funding agencies for their financial support. This project was supported by the National Institutes of Health R01NS079691 (TS) and the DermSTP Training Grant T32-AR058921 (DG).
References
- [1].a) Turturro MV, Christenson MC, Larson JC, Young DA, Brey EM, Papavasiliou G. PLoS One. 2013;8:e58897. doi: 10.1371/journal.pone.0058897. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Freytes DO, Wan LQ, Vunjak-Novakovic G. J Cell Biochem. 2009;108:1047–1058. doi: 10.1002/jcb.22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Martino MM, Mochizuki M, Rothenfluh DA, Rempel SA, Hubbell JA, Barker TH. Biomaterials. 2009;30:1089–1097. doi: 10.1016/j.biomaterials.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Brown AC, Rowe JA, Barker TH. Tissue Eng. Part A. 2011;17:139–150. doi: 10.1089/ten.tea.2010.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Markowski MC, Brown AC, Barker TH. Journal of Biomedical Materials Research Part A. 2012;100A:2119–2127. doi: 10.1002/jbm.a.34068. [DOI] [PubMed] [Google Scholar]; d Hodde J, Record R, Tullius R, Badylak S. Biomaterials. 2002;23:1841–1848. doi: 10.1016/s0142-9612(01)00310-6. [DOI] [PubMed] [Google Scholar]; e Gould ST, Darling NJ, Anseth KS. Acta Biomater. 2012;8:3201–3209. doi: 10.1016/j.actbio.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Lam J, Segura T. Biomaterials. 2013;34:3938–3947. doi: 10.1016/j.biomaterials.2013.01.091. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bryant SJ, Nuttelman CR, Anseth KS. J. Biomater. Sci.-Polym. Ed. 2000;11:439–457. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- [4].Gojgini S, Tokatlian T, Segura T. Mol Pharm. 2011;8:1582–1591. doi: 10.1021/mp200171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Burdick JA, Khademhosseini A, Langer R. Langmuir : the ACS journal of surfaces and colloids. 2004;20:5153–5156. doi: 10.1021/la049298n. [DOI] [PubMed] [Google Scholar]; b Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wong DY, Griffin DR, Reed J, Kasko AM. Macromolecules. 2010;43:2824–2831. [Google Scholar]; d Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS. Nat Mater. 2011;10:799–806. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- [6].Gu Z, Tang Y. Lab on a chip. 10:1946–1951. doi: 10.1039/c001335h. [DOI] [PubMed] [Google Scholar]
- [7].Dallabrida SM, Falls LA, Farrell DH. Blood. 2000;95:2586–2592. [PubMed] [Google Scholar]
- [8].a) Schense JC, Hubbell JA. Bioconjugate chemistry. 1999;10:75–81. doi: 10.1021/bc9800769. [DOI] [PubMed] [Google Scholar]; b Zisch AH, Schenk U, Schense JC, Sakiyama-Elbert SE, Hubbell JA. Journal of controlled release : official journal of the Controlled Release Society. 2001;72:101–113. doi: 10.1016/s0168-3659(01)00266-8. [DOI] [PubMed] [Google Scholar]; c Ehrbar M, Rizzi SC, Schoenmakers RG, Miguel BS, Hubbell JA, Weber FE, Lutolf MP. Biomacromolecules. 2007;8:3000–3007. doi: 10.1021/bm070228f. [DOI] [PubMed] [Google Scholar]; d Hu BH, Messersmith PB. Journal of the American Chemical Society. 2003;125:14298–14299. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]
- [9].Ichinose A, Tamaki T, Aoki N. FEBS Letters. 1983;153:369–371. doi: 10.1016/0014-5793(83)80645-0. [DOI] [PubMed] [Google Scholar]
- [10].Martino MM, Mochizuki M, Rothenfluh DA, Rempel SA, Hubbell JA, Barker TH. Biomaterials. 2009;30:1089–1097. doi: 10.1016/j.biomaterials.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Griffin DR, Kasko AM. Journal of the American Chemical Society. 2012:13103–13107. doi: 10.1021/ja305280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Griffin DR, Kasko AM. ACS Macro Letters. 2012;1:1330–1334. doi: 10.1021/mz300366s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Goodwin JF, Choi SY. Clin Chem. 1970;16:24–31. [PubMed] [Google Scholar]
- [14].a) McCray JA, Trentham DR. Annual Review of Biophysics and Biophysical Chemistry. 1989;18:239–270. doi: 10.1146/annurev.bb.18.060189.001323. [DOI] [PubMed] [Google Scholar]; b Walker JW, Reid GP, McCray JA, Trentham DR. Journal of the American Chemical Society. 1988;110:7170–7177. [Google Scholar]
- [15].Lei Y, Gojgini S, Lam J, Segura T. Biomaterials. 2011;32:39–47. doi: 10.1016/j.biomaterials.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ng YS, Rohan R, Sunday ME, Demello DE, D'Amore PA. Developmental Dynamics. 2001;220:112–121. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1093>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- [17].a) McDougall S, Dallon J, Sherratt J, Maini P. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2006;364:1385–1405. doi: 10.1098/rsta.2006.1773. [DOI] [PubMed] [Google Scholar]; b Steed DL. Surgical Clinics of North America. 1997;77:575–586. doi: 10.1016/s0039-6109(05)70569-7. [DOI] [PubMed] [Google Scholar]
- [18].Balkwill F. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- [19].Tokunaga A, Oya T, Ishii Y, Motomura H, Nakamura C, Ishizawa S, Fujimori T, Nabeshima Y, Umezawa A, Kanamori M, Kimura T, Sasahara M. J Bone Miner Res. 2008;23:1519–1528. doi: 10.1359/jbmr.080409. [DOI] [PubMed] [Google Scholar]
- [20].Owen SC, Fisher SA, Tam RY, Nimmo CM, Shoichet MS. Langmuir. 2013;29:7393–7400. doi: 10.1021/la305000w. [DOI] [PubMed] [Google Scholar]
- [21].Wong DY, Griffin DR, Reed J, Kasko AM. Macromolecules. 43:2824–2831. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.