

Abstract
PDZ (PSD-95, DiscsLarge, ZO1) domains function in nature as protein binding domains within scaffold and membrane-associated proteins. They comprise ~ 90 residues and make specific, high affinity interactions with complementary C-terminal peptide sequences, with other PDZ domains, and with phospholipids. We hypothesized that the specific, strong interactions of PDZ domains with their ligands would make them well suited for use in affinity chromatography. Here we describe a novel affinity chromatography method applicable for the purification of proteins that contain PDZ domain-binding ligands, either naturally or introduced by genetic engineering. We created a series of affinity resins comprised of PDZ domains from the scaffold protein PSD-95, or from neuronal nitric oxide synthase (nNOS), coupled to solid supports. We used them to purify heterologously expressed neuronal proteins or protein domains containing endogenous PDZ domain ligands, eluting the proteins with free PDZ domain peptide ligands. We show that Proteins of Interest (POIs) lacking endogenous PDZ domain ligands can be engineered as fusion products containing C-terminal PDZ domain ligand peptides or internal, N- or C-terminal PDZ domains and then can be purified by the same method. Using this method, we recovered recombinant GFP fused to a PDZ-domain ligand in active form as verified by fluorescence yield. Similarly, chloramphenicol acetyltransferase (CAT) and β-Galactosidase (LacZ) fused to a C-terminal PDZ domain ligand or an N-terminal PDZ domain were purified in active form as assessed by enzymatic assay. In general, PDZ domains and ligands derived from PSD-95 were superior to those from nNOS for this method. PDZ Domain Affinity Chromatography promises to be a versatile and effective method for purification of a wide variety of natural and recombinant proteins.
Keywords: HaloTag, PDZ Domain, Affinity Chromatography, NMDA Receptor, synGAP, Neuronal
Introduction
PDZ domains are ubiquitous, small (~90 residue), compact, modular protein-binding domains that hold together and organize membrane-associated signal transduction complexes [1-4]. They are often found in multi-domain scaffolding proteins that link together large molecular complexes at specific locations within cells. PDZ domains contain a ligand binding pocket that binds short peptide motifs at the C-termini of partner proteins [5, 6]. Some have also been shown to bind phospholipids and other PDZ domains [7, 8]. They can be divided into three distinct classes based on their specificity for distinct C-terminal peptide sequences [3, 5]. Type I PDZ domains bind C-terminal X-S/T-X-V/I/ L peptide motifs, where X denotes any amino acid. Type II PDZ domains bind C-terminal X-Φ-X-Φ peptide motifs, where Φ denotes the bulky, hydrophobic residues V/Y/F/L/I. Type III PDZ domains bind C-terminal X-D/E-X-V/ L peptide motifs. The affinities of PDZ domains for their ligands range from 0.1 to 10 μM [5, 9-15].
In addition to binding C-terminal peptide motifs, certain PDZ domains, such as the PDZ domain of neuronal nitric oxide synthase (nNOS), can bind homotypically to other PDZ domains via a β-Hairpin that immediately follows the core domain. The β-hairpin structure of nNOS binds to the ligand binding pocket of PDZ2 from PSD-95 or to the PDZ domain of syntrophin [16] to form a complex in which the binding pocket of the nNOS PDZ domain remains unoccupied and capable of binding to a PDZ domain ligand from another protein [17, 18].
Because of the specificity and high affinity of PDZ domains for their C-terminal ligands and for PDZ domains containing β-hairpin ligands, we reasoned that these interacting pairs could provide the core components of an affinity chromatography system including affinity resins, affinity tags, and elution agents. Previously, the PDZ1 domain of the Drosophila InaD protein was linked to a support resin and used to purify proteins engineered to contain the NorpA PDZ1 peptide ligand [19]. This ligand includes a cysteine residue that attaches covalently via a disulfide bond to the InaD PDZ1 binding pocket. Proteins were eluted from the InaD affinity column by exposure to a reducing reagent. Here we show that affinity columns containing the PDZ domains of PSD-95 can be used to purify active PDZ domain-binding proteins to very high purity in a single step without disulfide bond formation. We prepared solid supports derivatized with recombinant PDZ domains from PSD-95 and used them to purify five heterologously expressed neuronal proteins that contain endogenous PDZ domain ligands (Table 1), eluting them with synthetic peptides having the sequences of cognate PDZ domain ligands.
Table 1. Heterologously Expressed Neuronal Proteins Containing Endogenous PDZ Domain Ligands.
| Name | Organism | Residues | N-terminal Tag | PDZ Domain Resin | Elution Peptide |
|---|---|---|---|---|---|
| GluN2B Tail | Mus musculus | 842-1482 | MBP | PDZ2 | SIESDV |
| r-synGAP | Rattus norvegicus | 103-1293 | His Tag | PDZ3 | YKQTSV |
| CRIPT | Rattus norvegicus | Full Length | - | PDZ3 | YKQTSV |
| nNOS PDZbh | Mus musculus | 11-129 | - | PDZ2 | SIESDV |
| cypin | Homo sapiens | Full Length | MBP or His Tag | PDZ1+PDZ2 | SIESDV |
We also show that addition of PDZ domain-related affinity tags to POIs that do not contain endogenous PDZ domains or ligands enables purification of the POIs on the affinity resins. We used peptides derived from the C-terminal PDZ domain ligand of the N-methyl-D-aspartate type glutamate receptor (NMDAR) subunit GluN2B, PDZ domains from PSD-95, and the nNOS PDZ β-Hairpin domain (PDZbh) to construct affinity tags and corresponding affinity resins. We verified that the tags do not alter protein activity by fusing PDZ domain-derived affinity tags to DasherGFP, β-Galactosidase (LacZ), and chloramphenicol acetyltransferase (CAT) and then assaying their activity using standardized spectrophotometric and fluorescence assays.
Materials and Methods
Bacterial Strains and Materials
TOP10 cells (Cat. No. C4040-10, Life Technologies, Carlsbad CA) were used for plasmid DNA propagation and cloning, and BL21(DE3) cells (Cat. No. 69450-4l, EMD Millipore, Billerica MA) for protein expression. PfuUltra II Fusion HS DNA Polymerase (Cat. No. 600670, Agilent, Santa Clara CA) was used for Polymerase Incomplete Primer Extension-Ligation Independent Cloning (PIPE-LIC) and for Polymerase Incomplete Primer Extension Mutagenesis (PIPE-Mutagenesis) [20, 21]. All restriction enzymes (PvuI, Cat. No. R3150S; SbfI, Cat. No. R3642S) and DNA ligase (QuickLigase, Cat. No. M2200S) were purchased from New England Biolabs (Ipswitch MA). Chloramphenicol (Cat. No. C0378), acetyl-CoA (Cat. No. A2056), 5,5’-dithiobis-2-nitrobenzoic acid (DTNB) (Cat. No. D8130), CAT enzyme (Cat. No. C8413), o-nitrophenol-β-galactoside (ONPG) (Cat. No. N1127), o-nitrophenol (ON) (Cat. No. N19702), reducing agents tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (Cat. No. C4706) and β-mercaptoethanol (Cat. No. M6250), and buffers were purchased from Sigma Aldrich (St. Louis MO). Peptides used for elution of bound protein from affinity resin (YKQTSV and SIESDV) or for coupling to affinity resin (GAGSSIESDV) were purchased from Genscript (Piscataway NJ).
Expression Vectors and Cloning
The following cDNAs were codon-optimized for expression in E. coli, synthesized, and inserted into the pJExpress414 expression vector (Cat. No. pJ414) by DNA 2.0 (Menlo Park CA): Mus musculus GluN2B (Residues 842 to 1482; Q01097) fused to maltose binding protein (MBP-GluN2B Tail); E. coli thioredoxin (THX) (P0AA25); Dasher Green Fluorescent Protein (DasherGFP; Custom Order, DNA2.0, Menlo Park CA); Rattus norvegicus CRIPT (Q792Q4); residues 11 to 129 of Mus musculus nNOS (Q9Z0J4), containing the PDZ domain (single PDZbh) or two fused copies of the PDZ domain (tandem PDZbh); and PDZ1 (residues 61 to 151), PDZ2 (residues 155 to 249), PDZ3 (residues 302 to 402) and PDZ1-2 (residues 61 to 249) of Mus musculus PSD-95 (Q62108). A cDNA encoding MBP in pMAL-c4e plasmid (Cat. No. 8110) was purchased from New England Biolabs (Ipswitch MA) and a cDNA encoding glutathione S-transferase (GST) in pGEX-6P-1 plasmid (Cat. No. 28-9546-48) was purchased from General Electric Healthcare (Uppsala, Sweden). The two were transferred into pJExpress414 plasmids containing protein domains via PIPE-LIC to generate expression vectors encoding the proteins fused to MBP, GST or MBP-GST tags. A cDNA encoding E. coli K12 β-Galactosidase (LacZ) (P00722) in pMS34 plasmid (Cat. No. 32297) was purchased from AddGene (Cambridge MA). A cDNA encoding CAT (R0JP21/Q5YFS3/Q4VY50) in pCAT3-Basic plasmid (Cat. No. E1871) was purchased from Promega (Madison WI). cDNAs encoding Rattus norvegicus synGAP (Q9QUH6) and Homo sapiens cypin (Q9Y2T3) in pCMV6 plasmids (Cat. No. RN200179 and RC208771, respectively) were purchased from OriGene (Rockville MD) and were transferred into pJExpress414 expression vectors containing an N-terminal 6xHis Tag (synGAP residues 103 to 1293), MBP Tag (cypin) or no tag (cypin) via PIPE-LIC.
To generate expression vectors for the production of PDZ domain-HaloTag fusion proteins, genes encoding the PDZ domains from PSD-95 (PDZ1, PDZ2, PDZ3, PDZ1-2) and nNOS (PDZbh) were excised from pJExpress414 with PvuI and SbfI restriction enzymes, separated by agarose electrophoresis, gel-purified and ligated into gel-purified pFN18A (Cat. No. G2751, Promega, Madison WI) digested with PvuI and SbfI.
To generate affinity tags derived from C-terminal peptide PDZ domain ligands, cDNAs encoding fusion proteins with an N-terminal GST, MBP or THX were cloned into pJExpress414 in frame to DNA encoding the C-terminal ten amino acids of GluN2B (EKLSSIESDV), termed GluN2B ligand, by PIPE-LIC. Specific truncations of zero to ten amino acids of the GluN2B ligand fused in frame to an N-terminal GST, MBP, or THX were generated by PIPE-Mutagenesis.
To generate internal, N-terminal, or C-terminal PDZ domain or PDZbh domain affinity tags, cDNAs encoding the PDZ2 domain from PSD-95, or single or tandem PDZbh domains from nNOS, were cloned into pJExpress414 plasmids in-frame with cDNAs encoding MBP, GST or MBP-GST fusion proteins via PIPE-LIC. PDZ domain affinity tags were inserted N-terminally or C-terminally into MBP, or internally into the MBP-GST fusion protein.
cDNAs encoding DasherGFP (in pJExpress414), CAT (in pCAT3-Basic) and LacZ (in pMS34) were fused in frame with cDNA encoding the GluN2B ligand affinity tag, or the N-terminal PDZ2 affinity tag, or the PDZbh affinity tag into pJExpress414 plasmids by PIPE-LIC cloning.
Expression of Recombinant Proteins
Single colonies of BL21(DE3) cells harboring a pJExpress414 or pFN18A plasmid were grown overnight at 37 °C in 5 ml of lysogeny broth (LB) (Cat. No. L9110, Teknova, Hollister CA) supplemented with 100 μg/ml carbenicillin. Unless specified otherwise, overnight cultures were diluted 1:500 into Overnight Express Instant Terrific Broth (TB) Media (Cat. No. 71491, EMD Millipore, Billerica MA), grown for 16 hours at 37 °C, pelleted by centrifugation, and flash frozen in liquid nitrogen.
Cells expressing synGAP or MBP-GluN2B ligand were grown in LB medium at 37 °C until cultures reached an O.D.600 of 0.8. Cultures were then chilled to 18°C, and IPTG was added to a final concentration of 0.2 or 1 mM, for synGAP or the MBP-GluN2B ligand, respectively. Cultures were grown for an additional 24 hours at 18 °C.
Synthesis of PDZ Domain-HaloTag-HaloLink Affinity Resin
We describe the synthesis of 10 ml of PDZ Domain-HaloTag-HaloLink resin. This procedure was followed for synthesis of affinity resins linked to PDZ1, PDZ2, PDZ1 plus PDZ2, and PDZ3 from PSD-95, and for affinity resins linked to single and tandem PDZbh. Bacterial cell pellets (~10 grams) containing PDZ domain-HaloTag fusion proteins were resuspended in 10 ml of Purification Buffer [50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM TCEP, 0.5 mM EDTA, 10 mM MgCl2, 2 mM phenylmethylsulfonyl fluoride (PMSF) (Cat. No. 78830, Sigma Aldrich, St. Louis MO), Complete Protease Inhibitor (Cat. No. 04693116001, Roche, Indianapolis IN)], supplemented with 25 U/ml Benzonase (Cat. No. 71206, EMD Millipore, Billerica MA) and 10 U/ml ReadyLyse (Cat. No. R1810M, Epicentre, Madison WI), per gram of cells. Cells were evenly suspended in a Teflon-glass homogenizer and then lysed by three passes through a ML-110 microfluidizer (Microfluidics, Westwood MA). The cell lysate was clarified by centrifugation at 30,000 × g for 60 minutes at 4°C. The clarified lysate was added to 10 ml of settled HaloLink resin (Cat. No. G1915, Promega, Madison WI) that had been pre-equilibrated in Purification Buffer and then mixed with continuous agitation for 1.5 hours at 4°C on an end-over-end mixer. Unbound protein was separated from the derivatized resin by centrifugation at 2,000 × g for 5 minutes at 4°C. The PDZ-HaloTag-HaloLink resin was then resuspended in 1 column volume of Purification Buffer and transferred to a Glass Econo-Column Chromatography Column (Cat. No. 737-0717, BioRad, Hercules CA), capped, and allowed to settle for 60 minutes before the cap was removed. The resin was washed with 20 column volumes of Purification Buffer and then with 20 column volumes of Purification Buffer supplemented with 0.05% NaN3. It was resuspended in 1 column volume of Purification Buffer supplemented with 0.05% NaN3, and stored at 4°C.
To determine the density of PDZ domains on the resin, we released the PDZ domains from the resin by digestion of 200 μl of settled resin with ProTEV Plus (Cat. No. V6102, Promega, Madison WI). We determined the amount of protein released by SDS-PAGE as described below under “Assessment of Protein Purity and Yield.” The densities varied from 45 to 350 pmol PDZ domain/μl resin (Table 2).
Table 2. Ligand Densities of PDZ Domain-HaloTag-HaloLink Affinity Resins.
Ligand densities on the resin were measured as described in Methods.
| PDZ Domain Resin | Ligand Density (pmol PDZ Domain / μl Resin) |
|---|---|
| PDZ1+PDZ2 | 55 |
| PDZ1 | 65 |
| PDZ2 | 53 & 266 |
| PDZ3 | 350 |
| Single PDZbh | 89 |
| Tandem PDZbh | 45 |
Synthesis of GluN2B ligand-NHS-Agarose Affinity Resin
We describe the synthesis of 10 ml of GluN2B ligand-NHS-Agarose. Twenty ml of a 50% slurry of N-hydroxysuccinimide (NHS)-Activated Agarose Resin in acetone storage solution (Cat. No. 26200, Pierce, Rockford IL) was added to a 50 ml Falcon Tube. The storage solution was removed by centrifugation at 1,000 × g for 1 minute. The 10 ml of settled NHS-Activated Agarose resin was washed twice with 3 column volumes of ultrapure water followed by centrifugation at 1,000 × g for 1 minute. The NHS-Activated Agarose was then washed twice with 3 column volumes of Coupling/Wash Buffer (100 mM NaHPO4, pH 7.2, 150 mM NaCl) followed by centrifugation at 1,000 × g for 1 minute. Peptide Buffer (2.5 column volumes of 100 mM NaHPO4, pH 7.2, 150 mM NaCl, 10 mg/ml GAGSSIESDV peptide) was added to the resin, and the mixture was incubated for 2 hours at room temperature with continuous agitation on an end-over-end mixer. The GluN2B ligand-NHS-Activated Agarose was pelleted by centrifugation at 1,000 × g for 1 minute, and washed twice with 3 column volumes of Coupling/Wash Buffer followed by centrifugation at 1,000 × g for 1 minute. The remaining active sites on the GluN2B ligand-NHS-Agarose were blocked by incubation with 3 column volumes of Quenching Buffer (1 M ethanolamine, pH 7.4) for 20 minutes at room temperature with end-over-end mixing. Quenching Buffer was removed by centrifugation at 1,000 × g for 1 minute. GluN2B ligand-NHS-Agarose resin was then washed twice with 4 column volumes of Coupling/Wash Buffer separated by centrifugation at 1,000 × g for 1 minute. For storage, the GluN2B ligand-NHS-Agarose was washed twice with 4 column volumes of Storage Buffer (100 mM NaHPO4, pH 7.2, 150 mM NaCl, 0.05% NaN3), pelleted by centrifugation at 1,000 × g for 1 minute, resuspended in 1 column volume of Storage Buffer, and stored at 4°C.
Coupling of peptide to NHS-Agarose and the ligand density of immobilized peptide were monitored by the 660 nm Protein Assay (Cat. No. 22660, Pierce, Rockford IL) (The NHS leaving group interferes with BCA assays and absorbance assays at 280 nm). Ligand densities varied from 20 to 26 nmol peptide/μl resin (20-26 mM), which represents coupling efficiencies of 74 to 96%, respectively.
Purification of PDZ2 Domain of PSD-95 and Synthesis of PDZ Domain-NHS-Agarose Affinity Resin
We describe the purification of the PDZ2 domain of PSD-95 and the synthesis of 5 ml of PDZ domain-NHS-Agarose resin. The PDZ2 domain of PSD-95 was expressed in E. coli as described above and purified by chromatography on GluN2B ligand-NHS-Agarose Affinity Resin (synthesis described above). Bacterial cell pellets (~20 grams) containing recombinant PDZ2 domains were resuspended in 10 ml of Purification Buffer, supplemented with 25 U/ml Benzonase and 10 U/ml ReadyLyse, per gram of cells. Cells were evenly suspended in a Teflon-glass homogenizer and then lysed by three passes through a microfluidizer. The cell lysate was clarified by centrifugation at 30,000 × g for 60 minutes at 4°C. The clarified lysate was added to 15 ml of settled GluN2B ligand-NHS-Agarose that had been pre-equilibrated in Purification Buffer, and the suspension was incubated for 1 hour at 4°C with continuous agitation on an end-over-end mixer. Unbound protein was separated from the affinity resin by centrifugation at 2,000 × g for 2 minutes at 4°C. The resin was resuspended in 1 column volume of Purification Buffer and transferred to a Glass Econo-Column Chromatography Column, capped, and allowed to settle for 60 minutes before the cap was removed. The resin was washed with 20 column volumes of Purification Buffer. The PDZ2 domain protein was eluted by application of 4 column volumes of Peptide Elution Buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM TCEP, Complete Protease Inhibitor, and 400 μg/ml SIESDV peptide). Aliquots (17.5 ml) of eluted PDZ2 domain protein were pipetted into 20 ml Pierce 9K Protein Concentrators (Cat. No. 89885A, Thermo Scientific, Rockford IL) which were then subjected to centrifugation at 3,000 × g for 25 minutes at 4°C in a swinging bucket rotor. The filtrate was discarded, and another 17.5 ml aliquot of eluted PDZ2 domain protein was added to the concentrators and subjected to centrifugation. These steps were repeated until the final total volume of PDZ2 domain protein was reduced to 10 ml. The concentrated PDZ2 domain pool (11.3 mg/ml, 723 μM) was subjected to 5 cycles of buffer exchange into Coupling/Wash Buffer lacking free peptide in preparation for coupling to NHS-Activated Agarose. The five cycles of buffer exchange also served to remove bound GluN2B peptide from the purified PDZ2 domains.
Purified PDZ2 domains in Coupling/Wash Buffer (11.3 mg/ml, 723 μM) were coupled to NHS-Activated Agarose resin by the same procedure described for coupling of the GluN2B ligand to NHS-Activated Agarose resin. The density of PDZ2 domains on the resin was approximately 0.724 nmol protein/μl resin (724 μM; 9.4 mg/ml).
Protein Purification on PDZ2, PDZ1 plus PDZ2, and PDZ3-HaloTag-HaloLink Resin, Single and Tandem PDZbh Resin, PDZ2-NHS-Agarose and GluN2B ligand-NHS Agarose Resin
Unless otherwise specified, all bacterial cell pellets (0.4 - 2.5 grams per purification) containing tagged or untagged recombinant POIs were resuspended in 5 ml/g cell paste of BugBuster Lysis Buffer [1x BugBuster (Cat. No. 70921, EMD Millipore, Billerica MA), 50 mM HEPES, 150 mM NaCl, 5 mM TCEP, 0.5 mM EDTA, 10 mM MgCl2, 2 mM PMSF, Complete Protease Inhibitor supplemented with 25 U/ml Benzonase and 10 U/ml ReadyLyse]. Resuspended cells were incubated on a shaking platform at low speed for 20 minutes at room temperature before the lysate was clarified by centrifugation at 16,000 × g for 30 min at 4°C.
All protein purification was performed by gravity-flow, batch chromatography. Clarified lysate (1.8 to 14 ml) containing the POI was added to 100-200 μl of resin pre-equilibrated with Purification Buffer, and the suspension was agitated at 4 °C for 1 hour on an end-over-end mixer to allow the protein to bind to the affinity resin. The mixtures were then subjected to centrifugation at 1,000 × g for 2 minutes, and the supernatant was removed by pipetting. The affinity resin with bound protein was then resuspended in 2 column volumes of Purification Buffer, transferred to a BioSpin Chromatography Column (Cat. No. 732-6008, BioRad, Hercules CA), capped, and allowed to settle for 15 minutes. The cap was removed and the resin washed with 20 column volumes of Purification Buffer. Bound protein was eluted by application of 4 column volumes of Peptide Elution Buffer [50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM TCEP, Roche Complete Protease Inhibitor, and 400 μg/ml YKQTSV peptide (for PDZ3 Domain-resin) or SIESDV peptide (for PDZ2 Domain-resin, PDZ1 plus PDZ2 Domain-resin, and single and tandem PDZbh Domain-resin)]. The concentration of the eluted protein could be increased by decreasing the volume of buffer added for elution, increasing the concentration of peptide from 400 to 800 μg/ml, and/or by incubating the column resin with Peptide Elution Buffer without flow for 20 minutes before collecting the eluate. Five cycles of buffer exchange using an Amicon Ultra-0.5 ml concentrator and Peptide Elution Buffer lacking free peptide (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM TCEP, Roche Complete Protease Inhibitor) was sufficient to remove residual bound peptide remaining from the elution.
Remaining bound protein (<10% of bound protein before elution) and protein contaminants were removed from the resin by applying 4 column volumes of Denaturing Buffer (8 M Urea). The resin was then washed with 15 column volumes of Storage Buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM TCEP, 0.05% NaN3) to renature the immobilized PDZ domains or GluN2B ligand. Regenerated resin was stored in BD Falcon Tubes (Cat. No. 352070, BD Biosciences, Bedford MA) at 4°C. No loss of binding capacity was observed after 5 cycles of elution, denaturation and renaturation or after storage for 6 months at 4°C.
Bacterial cell pellets containing the MBP-GluN2B ligand fusion protein, synGAP, and fusions of PDZ domains to functional proteins (DasherGFP, LacZ, and CAT) were resuspended in 5 ml/g cell-paste Purification Buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM TCEP, 0.5 mM EDTA, 10 mM MgCl2, 2 mM PMSF, Complete Protease Inhibitor) supplemented with 25 U/ml Benzonase and 10 U/ml ReadyLyse. The resuspended cells were sonicated (Branson, Danbury CT) 2x for 90 seconds/pass (15% power, 1.0 second on, 1.5 seconds off) before clarification by centrifugation at 16,000 × g for 30 min at 4°C. In addition, all buffers that we used for purification of synGAP contained 0.02% Tergitol Type NP-40 (Cat. No. NP40S, Sigma-Aldrich, St. Louis MO). Otherwise, the purification methods were as described for the other POIs.
Assessment of Protein Purity and Yield
We used SDS-PAGE to determine purity of proteins and to quantify yields throughout the purification process. Protein samples were diluted into 4x LDS buffer (Cat. No. NP0008, Life Technologies, Carlsbad CA) and heated to 70°C for 10 minutes before fractionation on NuPAGE Novex 4-12% Bis-Tris SDS-PAGE gradient gels (Cat. No. NP0008, Life Technologies, Carlsbad CA) run under reducing conditions with NuPAGE Antioxidant (Cat. No. NP0005), and MOPS or MES running buffers (Cat. Nos. NP0001 and NP0002), all purchased from Life Technologies (Carlsbad CA). Proteins in the gels were stained with Gel Code Blue (Cat. No. 24592, Thermo Scientific, Rockford IL), and imaged on a Licor Odyssey Imager (Licor Biosciences, Lincoln NE) at 700 nm. Molecular weights of stained proteins were verified by comparison to Precision Plus Protein All Blue Standards (Cat. No. 161-0373, BioRad, Hercules CA). The amount of each stained protein was measured in the gel by comparison to known amounts of bovine serum albumin (Cat. No. A5611), lysozyme (Cat. No. L6876) or LacZ (Cat. No. G8511), all purchased from Sigma-Aldrich (St. Louis MO). The protein standards were loaded onto each gel in lanes adjacent to the protein samples.
Assays of Protein Function
The integrity of the purified DasherGFP fusion protein’s fluorophore was assayed on a Hitachi F-4500 FL Fluorescence Spectrometer (Tokyo, Japan) at 22°C. Excitation (300-600 nm) and emission (350-700 nm) spectra were collected to verify fluorophore activity. The fraction of properly folded DasherGFP fusion proteins was measured by comparing the emitted fluorescence intensity at 520 nm of tagged, purified DasherGFP fusion proteins excited at 505 nm to that of an equimolar amount of untagged, purified DasherGFP.
The activity of purified LacZ fusion proteins was assayed by measuring the rate of hydrolysis of ONPG on a VERSAmax tunable microplate reader at 20°C as described in [22]. Briefly, the rate of ON release after addition of LacZ, produced by hydrolysis of ONPG, was measured by continuous monitoring of absorbance at 420 nm. Initial rates of ONPG hydrolysis were entered into Prism (v6.0d, GraphPad Software, La Jolla CA), plotted against ONPG substrate concentrations and analyzed by nonlinear regression using the Michaelis-Menten equation to calculate kcat and KM values. These values, calculated for purified LacZ fusion proteins, were compared to values from the literature [23] by ordinary one way ANOVA (Uncorrected Fisher’s LSD) in Prism. The purified LacZ fusion proteins were prepared for assay by buffer exchange in an Amicon Ultra-0.5 ml concentrator into 50 mM Tris, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.01 mM MnCl2 and 100 mM β-mercaptoethanol [24]. Aliquots were flash frozen in liquid nitrogen and stored at -80°C.
The activity of purified CAT fusion proteins was determined by measuring the rate of production of 5-thio-2-nitrobenzoic acid resulting from the reduction of DTNB by free CoA generated during acylation of chloramphenicol. The rate of reduction was measured by continuously monitoring absorbance at 412 nm after addition of CAT on a VERSAmax tunable microplate reader at 37 °C using a DTNB reduction assay described in [25]. Initial rates of chloramphenicol acylation were entered into Prism (v6.0d; GraphPad Software, La Jolla CA), plotted against acetyl-CoA or chloramphenicol substrate concentrations and analyzed by nonlinear regression using the Michaelis-Menten equation to calculate kcat and KM values. These values, calculated for purified CAT fusion proteins and for equimolar amounts of untagged CAT (Sigma), were compared by ordinary one way ANOVA (Uncorrected Fisher’s LSD) in Prism. The activity of purified CAT fusion proteins and equimolar amounts of untagged CAT (Sigma) were compared. CAT fusion proteins were prepared for assay by buffer exchange in an Amicon Ultra-0.5 ml concentrator into10 mM Tris, pH 7.8, 200 mM NaCl, 0.2 mM chloramphenicol, 0.1 mM β-mercaptoethanol [26]. Aliquots were flash frozen with liquid nitrogen and stored at -80°C.
Results and Discussion
Synthesis and Testing of Affinity Resins
Synthesis of PDZ Domain and PDZ Domain Binding Peptide Affinity Resin
Figure 1 outlines one possible workflow for PDZ Affinity Chromatography. A protein of interest (POI) containing a C-terminal PDZ domain peptide ligand is expressed in E. coli. The ligand can be endogenous or introduced by genetic engineering. Clarified bacterial lysate containing the POI is passed over a solid support resin derivatized with a recombinant PDZ domain. The PDZ domain ligand on the POI directs binding to the PDZ domain immobilized on the resin. After washing to remove loosely bound contaminants remaining from the cell lysate, free peptide ligand is added to the resin to competitively remove the bound POI from the immobilized PDZ domains, resulting in elution of a purified POI. In an alternative scheme, a POI containing a PDZ domain is passed over a solid support derivatized with a PDZ domain peptide ligand and eluted by addition of the free ligand (Fig. 2). In both of these schemes, recombinant affinity tags can be introduced with a TEV protease site between the tag and the POI, permitting removal of the tag by proteolysis to generate purified untagged protein.
Figure 1. Generalized PDZ Domain Affinity Chromatography Purification Scheme I: PDZ Domain Peptide Ligand Tag.
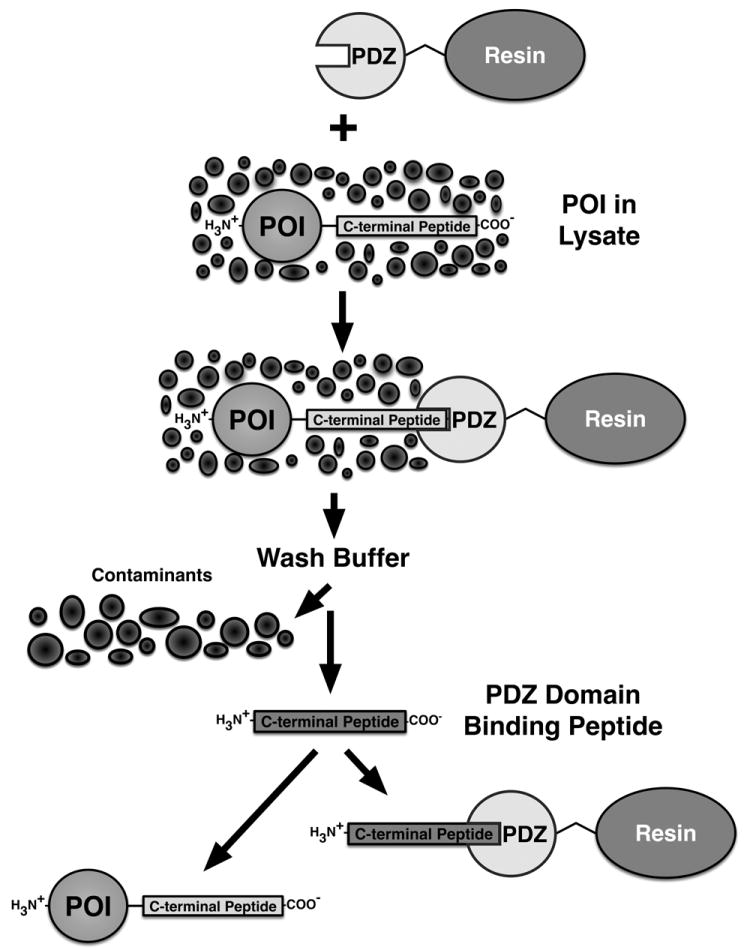
An expressed Protein Of Interest (POI), containing a naturally occurring or recombinantly appended PDZ Domain Peptide Ligand, can be captured from a cellular lysate by binding to a PDZ Domain coupled to a solid support resin. Extensive washing of the captured POI removes cellular contaminants. Free PDZ domain peptide ligand is then added to competitively elute the captured POI.
Figure 2. Generalized PDZ Domain Affinity Chromatography Purification Scheme II: PDZ Domain Tag.
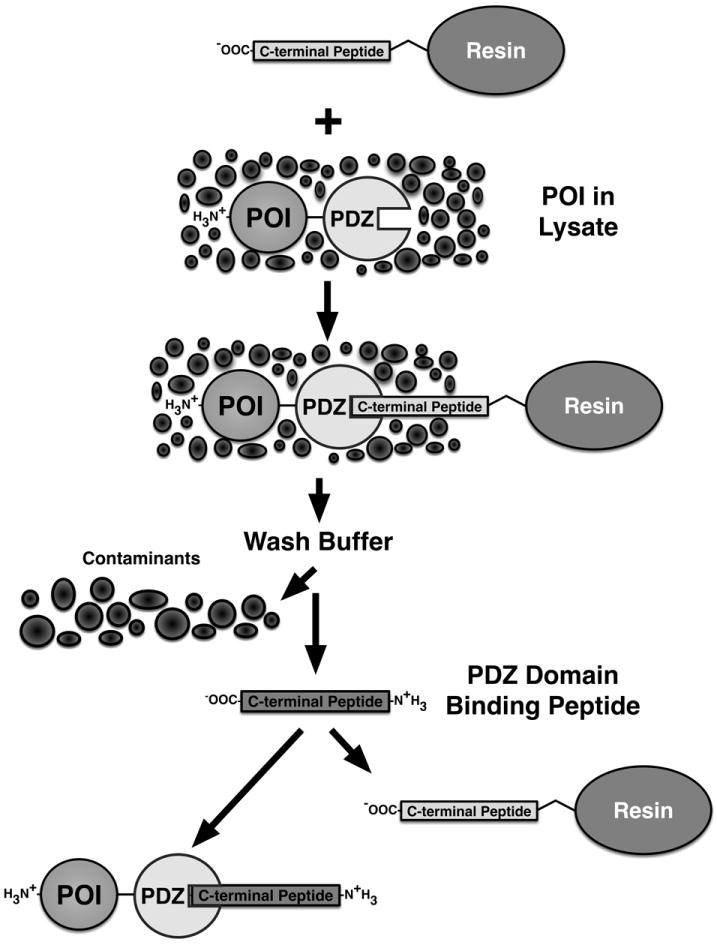
An expressed Protein Of Interest (POI) containing a naturally occurring or recombinantly appended PDZ domain can be captured from a cellular lysate by binding to a PDZ Domain-Peptide Ligand coupled to a solid support resin. Extensive washing of the captured POI removes cellular contaminants. As in Figure 1, free PDZ domain peptide ligand is then added to competitively elute the captured POI.
To prepare affinity resin containing immobilized PDZ domains, we chose to use the HaloTag and HaloLink resin manufactured by Promega because of its speed and ease of use compared to the conventional NHS coupling method, and also because it permits coupling with a single, uniform orientation. The HaloTag is a catalytically inactive bacterial haloalkane dehalogenase from Rhodococcus that has been engineered to rapidly form a covalent attachment to synthetic chloroalkane ligands [27, 28]. The HaloTag and HaloLink resin have been used for rapid, small scale preparation of affinity resins to purify polyclonal antibodies [29]. Using a similar procedure (described in Methods), we expressed HaloTag-PDZ domain fusion proteins in E. coli, lysed the cells, and incubated the clarified lysate with HaloLink resin to generate the PDZ domain-HaloTag-HaloLink affinity resin (Fig. 3). Coupling of PDZ Domain-HaloTag fusion proteins to HaloLink resin was monitored on SDS gels by determining the amount of PDZ domain protein released from the resin after cleavage of the HaloLink with TEV protease, as described in Methods. Coupling densities varied from 45 to 350 pmol PDZ domain/μl resin (Table 2).
Figure 3. Preparation of PDZ Domain-HaloTag-HaloLink Affinity Resin.
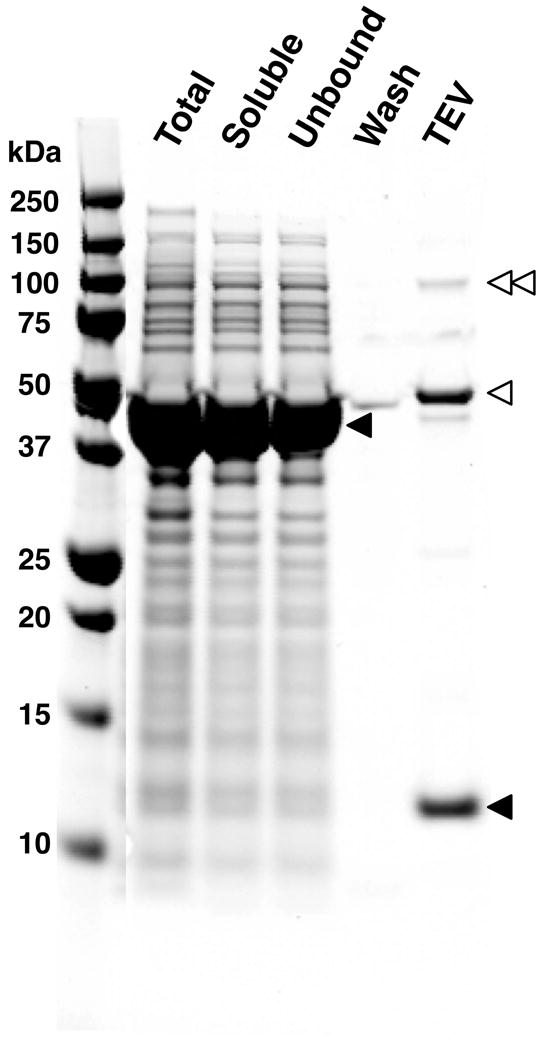
A fusion protein in which the PDZ3 domain was fused to the HaloTag was prepared as described in Methods. The PDZ3 Domain Affinity Resin was synthesized by coupling of the fusion protein to HaloLink Resin. The derivatized resin was washed extensively to remove contaminants. To measure the binding capacity of the resin, bound PDZ domains were released from an aliquot of the resin by digestion with TEV protease and quantified as described in Methods. In this experiment, resin capacity (pmol PDZ domain /μl resin) was 350 μM. Total protein, soluble protein, unbound protein, wash and TEV protease-treated fractions from the preparation were run on a 4-12% gradient SDS-PAGE gel with Precision Plus Protein All Blue Standards and stained with Gel Code Blue. Single and double unfilled arrowheads indicate the positions of monomeric and dimeric TEV protease, respectively. The upper and lower filled arrowheads indicate the positions of the PDZ3 domain-HaloTag fusion protein and the free PDZ3 domain, respectively. The lighter band below the TEV protease monomer is a contaminant present in the TEV protease.
In addition to preparing PDZ Domain Affinity Resin, we also prepared resin derivatized with PDZ domain peptide ligands for use in purifying POIs containing PDZ domains. Peptides were coupled to Agarose with an NHS linker, as described in Methods. The densities of peptide ligands on NHS coupled resin varied from 20 to 26 nmol peptide/μl resin.
Purification of the Cytosolic Domain of GluN2B
To test the utility of PDZ Domain Affinity Resin, we purified a panel of proteins known to interact with the PDZ domains of PSD-95 (Table 1). PSD-95 is a scaffold protein located in the postsynaptic density of glutamatergic synapses that contains three PDZ domains in addition to an SH3 and a guanylate kinase-like domain [1, 4]. PSD-95 is the first protein in which PDZ domains were recognized. Because of its critical role in receptor clustering and protein localization at the synapse [30-33], it is also one the most well characterized PDZ domain-containing proteins.
The N-methyl-D-aspartate type glutamate receptor (NMDAR) is one of three major classes of receptors for glutamate, the principal excitatory neurotransmitter in the central nervous system and has a seminal role in learning and memory through its actions as a “coincidence detector” that initiates changes in synaptic strength leading to the formation of new neural networks [34-37]. It is comprised of four subunits, including two from the GluN2 subfamily (GluN2A-D) [38]. The GluN2 subunits are unique among ligand-gated channels in having extended intracellular C-terminal domains or “tails,” which extend into the cytoplasm and serve as nucleation sites for scaffolds and signal transducing enzymes [39]. The cytoplasmic tails of NR2 subunits have not previously been expressed in soluble form in significant quantities, or purified. We chose to express and purify the cytoplasmic tail of the GluN2B subunit (“GluN2B Tail”: GluN2B residues 840-1482) which contains a C-terminal PDZ ligand that binds to PDZ2 of PSD-95. A PDZ Domain Affinity Resin substituted with PDZ2 from PSD-95 was synthesized as described in Methods. When expressed in E. coli, the GluN2B Tail forms insoluble protein aggregates known as inclusion bodies (Supp. Fig. 1A); however we found that it could be expressed in soluble form when fused at its N-terminus to MBP. The bacterial lysate containing the MBP-GluN2B Tail fusion protein was incubated with PDZ2 Domain Affinity Resin, and eluted with a competing peptide ligand as described in Methods. The eluted fusion protein was >70% pure (Fig. 4A) and was recovered at a concentration of 0.5 μM with a yield of 6% of the amount applied to the column. We found that PDZ Domain Affinity Chromatography was the method of choice for purifying the GluN2B Tail, compared to alternative methods. For example, after a similar column protocol substituting Amylose (MBP Tag) Affinity resin, the MBP-GluN2B Tail fusion protein was only approximately 8% pure and was recovered at a concentration of 0.1 μM with a yield of 6% of the amount applied to the column (Supp. Fig. 1B). Attempts to purify the MBP-GluN2B Tail fusion protein with Streptactin and Ni-NTA resins after addition of N- or C-terminal StrepII or 8xHis Tags, respectively, were unsuccessful; no protein was present in the column eluate (data not shown).
Figure 4. Purification of Heterologously Expressed Neuronal Proteins with Endogenous PDZ Domain Ligands.
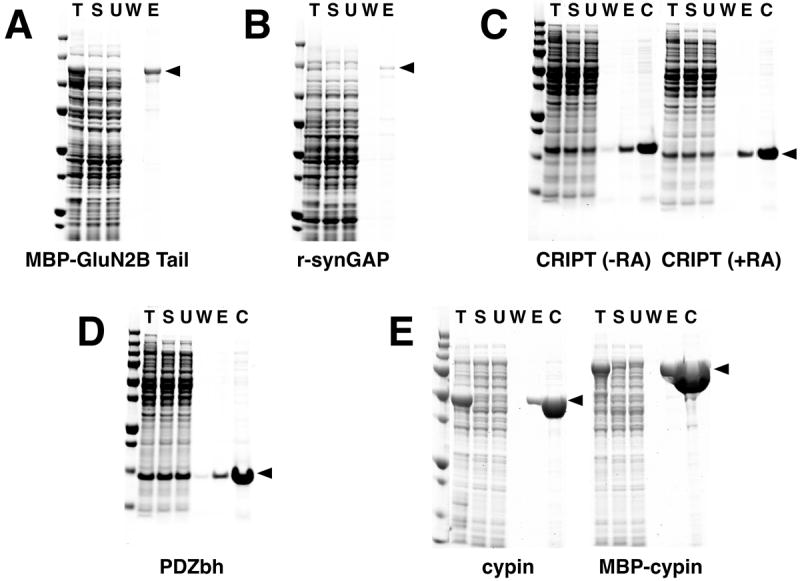
Six heterologously expressed neuronal proteins with naturally occurring PDZ domain ligands were purified on PDZ Domain Affinity Resins as described in Methods: (A) Purification of MBP-GluN2B Tail; (B) Purification of His-tagged r-synGAP; (C) Purification of CRIPT in the presence or absence of reducing agents (RA); (D) Purification of PDZbh; (E) Purification of cypin and MBP-cypin. MBP-GluN2B Tail and PDZbh were bound to PDZ2 Domain-HaloTag-HaloLink Resin and eluted with 400 μg/ml SIESDV peptide. Note that the ligand density of the PDZ2-resin used for purifications shown in (A) and (D) was 53 pmol/μl resin. Subsequent preparations of PDZ2-resin achieved the higher ligand density of 240-260 pmol/μl resin shown in Table 2. R-synGAP and CRIPT were bound to PDZ3 Domain-HaloTag-HaloLink Resin and eluted with 400 μg/ml YKQTSV peptide. Cypin and MBP-cypin were bound to tandem PDZ1+PDZ2 Domain-HaloTag-HaloLink Resin and eluted with 400 μg/ml SIESDV peptide. In the experiments shown in C and D the resin was incubated with an amount of recombinant POI in excess of the resin capacity. Fractions from each purification and Precision Plus Protein All Blue Standards were separated on a 4-12% gradient SDS-PAGE gel and stained with Gel Code Blue. T, Total protein; S, soluble protein; U, unbound protein; W, wash; E, peptide eluate; C, concentrated peptide eluate.
Purification of Soluble Recombinant SynGAP
SynGAP (Synaptic GTPase Activating Protein) is a dual Ras and Rap GTPase activating protein that is highly concentrated in the postsynaptic density of excitatory synapses [40]. Homozygous deletion of synGAP is lethal in mice [41, 42], and a heterozygous deletion confers behavioral phenotypes associated with cognitive disability and mental illness [43]. Synaptic plasticity is disrupted in heterozygous knockout mice and the formation of spine synapses during development is accelerated [42-44]. Mutations in the human SYNGAP1 gene appear to cause non-syndromic intellectual disability and certain forms of autism [45]. SynGAP is regulated by phosphorylation in a disordered region carboxyl to the GAP domain [46]. Attempts to understand the effects of phosphorylation have been hampered by the difficulty of purifying a soluble form of synGAP containing the disordered region. It was previously shown that removal of the N-terminal 102 residues of synGAP prevents the association with membranes of a recombinant form containing the PH, C2, and GAP domains (residues 103-725), but lacking the disordered region [47]. Therefore, we attempted to express and purify a soluble form of synGAP that also contains the disordered region beyond the GAP domain (r-synGAP; residues 103-1293). We found that r-synGAP remains soluble after expression in E. coli. This form of synGAP contains a carboxyl terminal ligand that binds to PDZ3 of PSD-95. Therefore, we synthesized PDZ3 Domain Affinity Resin and used it to purify r-synGAP to >70% homogeneity with a concentration of 0.2 μM and a yield of 6% of the soluble r-synGAP applied to the column (Fig. 4B), as described in Methods. When r-synGAP was tagged with an N-terminal 6x Histidine tag and purified by a similar procedure on Talon Affinity Resin, the purity of the recovered protein was less, approximately 25%, but the yield was considerably greater (75% at a concentration of 2 μM) than after PDZ3 Domain Affinity Chromatography (Supp. Fig. 1C). In addition to an appended N-terminal 6x Histidine tag, r-synGAP contains a naturally occuring internal 10x polyhistidine domain which markedly increases its affinity for Talon resin; we believe this contributes to its high yield from the Talon resin.
Purification of CRIPT (Cysteine Rich Interactor of PDZ Three)
CRIPT (Cysteine Rich Interactor of PDZ Three) is a small (101 residue), highly conserved, cysteine-rich protein concentrated in the PSD of glutamatergic synapses in pyramidal neurons [9, 48, 49]. CRIPT is one of three proteins known to bind to PDZ3 of PSD-95. It also binds the homologous PDZ3 domains of PSD-93 (chapsyn-110), SAP102 and SAP97 [9]. It can bind simultaneously to polymerized tubulin, thus linking PSD-95 family proteins to the microtubule cytoskeleton [48]. CRIPT contains 8 cysteine residues, all confined to CXXC repeats. To test whether PDZ3 Affinity Chromatography can be carried out in the presence of reducing agents, we chose to use it to purify recombinant CRIPT. CRIPT was previously purifed as a THX-fusion protein [48], so we attempted to purify it in the absence of exogenous affinity tags. We carried out the purification as described in Methods, except that, in one experiment, a reducing agent (5 mM TCEP) was present in all buffers; in a second the TCEP was not included. The results of the two purifications were identical. In the presence or absence of TCEP, CRIPT was purified to >95% homogeneity, with a yield of 7.6% and a concentration of 9 μM (Fig. 4C). In this case, the load of soluble CRIPT applied to the column exceeded the column capacity. Therefore, the yield was calculated as a percentage of the column capacity. Thus, unlike the NorpA-InaD affinity chromatography system [19], affinity chromatography with the PDZ domains from PSD-95 can be carried out either in the presence or absence of reducing agents.
Purification of PDZbh from nNOS (neuronal nitric oxide synthetase)
Neuronal nitric oxide synthase (nNOS) [50, 51] catalyzes the formation of nitric oxide and citrulline from arginine. Activation of nNOS by binding of Ca2+/calmodulin leads to formation of NO, which, in turn, activates guanylyl cyclase to form cGMP. NO generation can modulate synaptic plasticity by acting as a retrograde synaptic transmitter, and, in some circumstances provide neuroprotection against excitoxicity [52]. It contains a single PDZ domain (Residues 14-125 of Mus musculus nNOS) with an unusual structure that includes a β-Hairpin (bh) fold that itself acts as a PDZ domain ligand [17]. We were able to express the PDZbh domain from nNOS in E. coli and purify it to >95% homogeneity on the PDZ2 Domain Affinity Resin with a yield of 43%, as described in Methods (Fig. 4D). Again, the load of soluble POI applied to the column exceeded the column capacity. Therefore, the yield was calculated as a percentage of the column capacity. The concentration of purified PDZbh was 7.6 μM.
Purification of Cypin
Cypin was originally discovered and characterized as an abundant cytosolic protein that interacts directly with PSD-95 [53]. When overexpressed in neurons, it appears to trap PSD-95 in the cytosol and reduces the targeting of PSD-95 to synapses. During neural development, it contributes to regulation of dendritic branching, in part by interfering with the interaction between PSD-95 and microtubules [54]. Cypin binds to the PSD-95 family of proteins via a C-terminal SSSV sequence and has the unique property that it requires the presence of both PDZ1 and PDZ2 for detectable binding [53]. Cypin has been partially purified from brain extracts in small quantities by affinity purification with glutathione-beads bound to GST-PSD-95 [53]. More recently, both GST- and MBP- fusion proteins of cypin have been purified by standard affinity chromatography [55].
We used an affinity column substituted with a HaloTag-PDZ1+PDZ2 fusion protein (residues 61 to 249 of Mus musculus PSD-95) to purify heterologously expressed human cypin, as described in Methods. It was expressed in E. coli either as the free protein or fused to an N-terminal MBP, and purified from the bacterial supernate on the PDZ1+PDZ2 Domain-Affinity Resin, as described in Methods. Both cypin and MBP-cypin were purified to >99% homogeneity (Fig. 4E) with yields of 71% and 96% of the amount applied to the column, and concentrations of 0.5 μM and 0.8 μM, respectively.
In summary, we tested the utility of three different synthetic PDZ Domain Resins derivitized with PDZ domains from PSD-95 for the purification of five individual proteins. The tests were made by application of soluble bacterial lysates containing the expressed protein without individually optimizing the amount of protein loaded or the timing of incubation with the resin. MBP-GluN2B Tail and PDZbh of nNOS were purified on a PDZ2 Domain Resin. R-synGAP and CRIPT were purified on a PDZ3 Domain Resin. Finally, cypin and MBP-cypin were purified on a tandem PDZ1+PDZ2 Domain Resin. We successfully completed all of the purifications that we attempted. Yields of protein ranged from 6-8% for MBP-GluN2B, r-synGAP, and CRIPT, to 43-96% for PDZbh and Cypin. We suspect that the yields of the first three proteins could be improved by careful optimization. We found that no other commonly used affinity-resin and tag pairs worked for purification of MBP-GluN2B. Furthermore, CRIPT had not been previously purifed without a THX-affinity tag. CRIPT contains several labile cysteines; we were able to purify it in the presence of TCEP, demonstrating that the PDZ Domain Resins and their respective ligands are compatible with the use of reducing agents. Our results demonstrate that PDZ Domain Affinity Chromatography can be an excellent tool for the rapid purification of proteins with endogenous PDZ domain ligands.
Comparison of NHS and the HaloTag-HaloLink System for synthesis of PDZ Domain-Affinity Resin
We used the HaloTag-HaloLink system to synthesize most of the PDZ Domain Affinity Resins discussed here. However, we recognize that the cost of preparing HaloLink-HaloTag-PDZ domain resin would be prohibitive for large-scale applications in which yields of tens to hundreds milligrams of protein are required. Therefore, we tested a less expensive linkage method for preparation of the PDZ Domain Affinity Resins. We purified the recombinant PDZ2 domain from PSD-95 on a PDZ Domain Ligand Affinity Resin and coupled it to NHS-Agarose, as described in Methods (Fig. 5A). The purification yielded approximately 110 mg of 40 μM PDZ2 domain protein. The purified PDZ2 domain was concentrated to 723 μM in preparation for coupling to NHS-Agarose, as described in Methods. Residual PDZ Domain Peptide Ligand was then removed by five exchanges of buffer by ultrafiltration, each time diluting the concentrated protein with buffer that did not contain the peptide. The concentrated PDZ2 domain, freed of ligand, was coupled to NHS-Agarose, and unreacted NHS sites were quenched with ethanolamine as described in Methods. Coupling was monitored on SDS gels as described in Methods. The ligand density of PDZ2 domains on the coupled Agarose was 724 pmol/μl settled resin (724 μM; 9.4 mg/ml).
Figure 5. Comparison of HaloTag-HaloLink and NHS linkage for Synthesis of PDZ Domain-Agarose Affinity Resin.
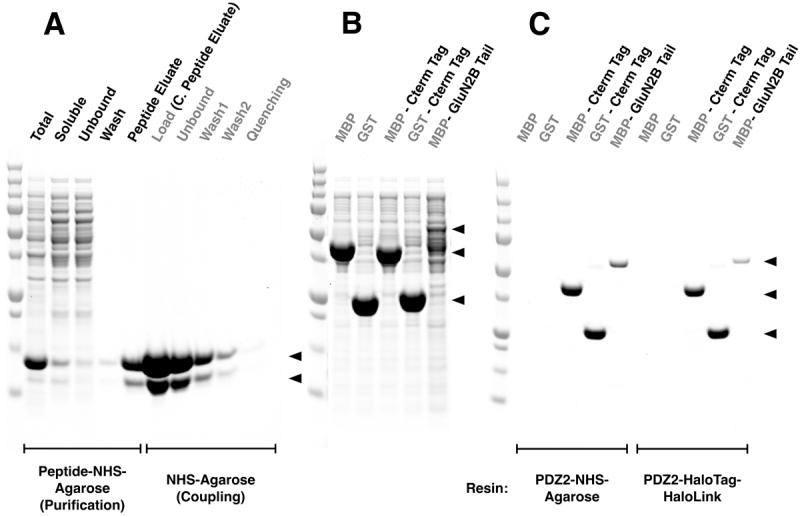
All samples and Precision Plus Protein All Blue Standards were fractionated by SDS-PAGE and protein was stained with Gel Code Blue.
(A) Purification of PDZ2 and coupling to NHS Agarose resin. The first 5 lanes show fractions from the purification of the PDZ2 domain on GluN2B ligand-NHS-Agarose as described in Methods: Total, bacterial extract; Soluble, extract after centrifugation; Unbound, column flow-through; Wash; Peptide Eluate, protein eluted with 400 μg/ml SIESDV peptide. The peptide was removed from the PDZ2 domain, as described in Methods, before linkage to NHS-Agarose. The next 5 lanes show fractions from coupling of the purified PDZ2 Domain to NHS-Agarose beads (as described in Methods): Load, concentrated peptide eluate that was incubated with NHS-Agarose; Unbound, supernatant after linkage; Wash1 & 2, two washes of the resin before quenching; Quenching, supernatant after quenching of unreacted NHS groups with ethanolamine. Upper and lower arrows indicate full length and truncated PDZ2 domain, respectively.
(B) Soluble lysate after heterologous expression of MBP, GST, their respective fusion proteins containing the 7-residue C-terminal PDZ2 Domain ligand Affinity Tag from GluN2B, and the entire GluN2B Tail fused to the C-terminus of MBP (see Methods).
(C) Purification of the fusion proteins shown in (B) on PDZ2-NHS-Agarose or on PDZ2 Domain-HaloTag-HaloLink resin. Soluble lysates were purified on the indicated Affinity Resins and eluted with 400 μg/ml SIESDV peptide, as described in Methods. The lysates containing MBP and GST fused to PDZ2 contained more protein than the capacity of the resin; thus these two columns were overloaded.
We compared PDZ2 Domain Affinity Resin coupled by NHS to that coupled by the HaloTag-HaloLink method by purifying a test panel of three proteins on each of the two resins: MBP-GluN2B Tail, and MBP and GST both fused to a PDZ2 Ligand Affinity Tag as described below and in Methods (Fig. 5B, C). All three proteins were purified to >95% homogeneity (Fig. 5C). We obtained higher amounts and higher percent recovery for MBP-GluN2B when the PDZ2 domain was linked to Agarose by NHS (1.2 μM [9.8%] vs. 0.5 μM [4.2%]). We also obtained larger amounts of MBP-PDZ2 ligand (12.9 μM vs. 9.6 μM) and GST- PDZ2 ligand (22.7 μM vs. 19.5 μM) from the NHS-linked resin. However, the yields of these two proteins, calculated as a percentage of the column capacity, was higher for the PDZ2-HaloTag-HaloLink resin: 16% vs 34% and 27% vs. 68%, respectively. The increased total recovery of protein for all of the POIs with NHS-coupled PDZ2 Domain Resin is likely due to its higher ligand density relative to the HaloLink-HaloTag-coupled resin (724 pmol/μl vs. 266 pmol/μl, respectively). Background binding to both resins was insignificant, as assessed by incubating them with untagged MBP and GST or with bacterial proteins in the E. coli lysate. These results indicate that PDZ domains can be successfully coupled to NHS-Agarose resin and that such resins work well for purification of proteins that contain PDZ domain ligands.
Engineering of Recombinant Affinity Tags into POIs
PDZ Domain C-terminal Peptide Ligands as Affinity Tags
The C-terminal six residues of the GluN2B subunit of the NMDAR (SIESDV) bind with high affinity to PDZ1 and PDZ2 of PSD-95, at KD’s of 2.3 and 0.7 μM, respectively [14]. We hypothesized that fusing the C-terminal residues of GluN2B to a POI would enable it to bind to the PDZ1 or PDZ2 domains of PSD-95 immobilized on an affinity resin. To test this hypothesis, we prepared recombinant proteins with varying numbers of residues from the GluN2B Tail (EKLSSIESDV) fused to GST, MBP or THX. To determine a minimum length of the ligand sequence required to permit purification of the proteins on PDZ2 Domain Affinity Resin, ten fusion proteins, containing one to ten of the ligand residues, were made for each of the three proteins. We found that the five C-terminal residues (IESDV) were all required for effective binding to, and for elution from, the PDZ2 Domain Affinity Resin (Fig. 6). GST fusion proteins with tags longer than 5 residues were purified to >95% homogeneity as assessed by SDS-PAGE (Fig. 6B). MBP and THX fused to the truncation library of one to ten residues were also purified to >95% homogeneity (Supp. Figs. 2 and 3). To minimize the possibility of structural interference in fusion proteins containing the PDZ2 Ligand Tag, we decided to use the seven residues from the C-terminus of GluN2B, SSIESDV, as a standard Ligand Tag.
Figure 6. PDZ Domain Peptide Ligand Affinity Tags: Fusion of GluN2B Peptide Ligand Truncation Library to GST.
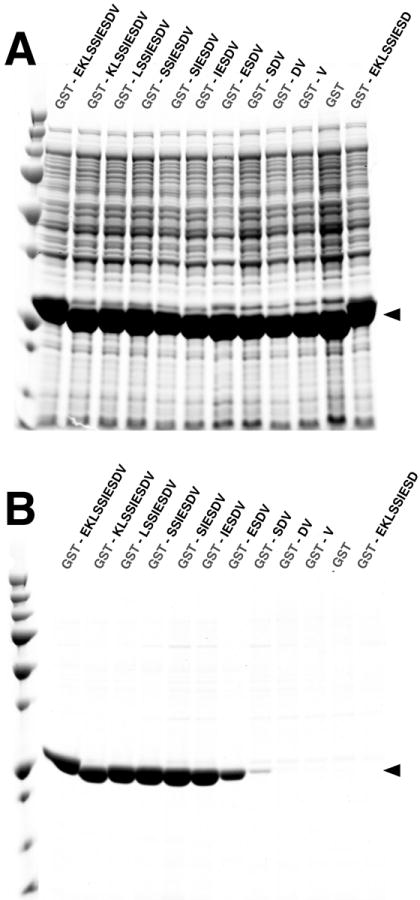
All samples and Precision Plus Protein All Blue Standards were fractionated on a 4-12% gradient SDS-PAGE gel and stained with Gel Code Blue.
(A) Soluble lysate after heterologous expression of Glutathione S-Transferase (GST) fused to a truncation library of the C-terminal ten residues of the GluN2B Tail.
(B) Glutathione S-Transferase (GST) fusion proteins shown in (A), after purification on a PDZ2 Domain-HaloTag-HaloLink resin and elution with 400 μg/ml SIESDV peptide.
PDZ domains as affinity tags
To assess the utility of conventional PDZ domains as affinity tags, we expressed the PDZ2 Domain of PSD-95 alone, fused N- or C-terminally to MBP, or fused internally between the MBP and GST proteins. Each fusion protein was purified with single or tandem PDZbh Domain Affinity Resins prepared with the HaloTag-HaloLink system, and with PDZ2 Domain Ligand Affinity Resin prepared with NHS-Agarose, as described in Methods (Fig. 7A and B). All of the PDZ2 Domain Tagged POIs, regardless of the position of the tag, were purified to >95% purity on all three types of resin (Fig. 7B). The yields of pure protein were higher when the tags were fused C-terminally or internally than when they were fused N-terminally. Yields of tagged protein were 25- to 120-fold higher for the PDZ2 Domain Peptide Ligand Affinity Resin than for single and tandem PDZbh Domain Affinity Resins: PDZ2 domain alone (36.9 μM vs. 0.8 and 0.3 μM); MBP-PDZ2 domain fusion (30.6 μM vs. 1.3 and 0.5 μM); and MBP-PDZ2 domain-GST fusion (19.1 μM vs. 0.8 and 0.5 μM). Control MBP or GST, expressed alone without a PDZ domain, showed no detectable binding to the Affinity Resins. This data demonstrates that PDZ Domain Peptide Ligand Affinity Resins may be particularly useful for a wide range of recombinantly tagged POIs. The utility of PDZ Domain Peptide Ligand Affinity Resins is enhanced by their low cost and ease of preparation compared to PDZbh Domain Affinity Resins.
Figure 7. PDZ Domain and PDZ Domain-binding Beta Hairpin (PDZbh) Affinity Tags.
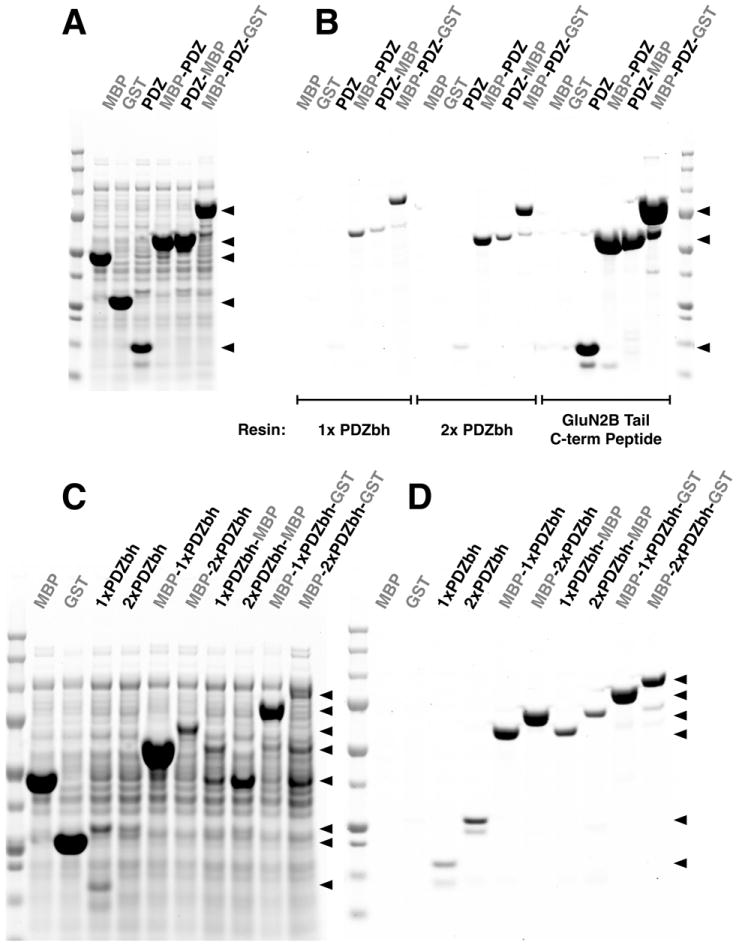
All samples and Precision Plus Protein All Blue Standards were run on a 4-12% SDS-PAGE gradient gel and stained with Gel Code Blue.
(A) Soluble lysates after heterologous expression of MBP, GST, a PDZ2 domain, MBP fusion proteins containing an N-terminal or C-terminal PDZ2 Tag, and an MBP-GST fusion protein containing an internal PDZ2 Domain Tag.
(B) Peptide eluates from purifications of the proteins shown in (A) on HaloTag-HaloLink Affinity Resins containing one (1x PDZbh) or two (2x PDZbh) PDZbh Domains, and on PDZ2 Domain Peptide Ligand-NHS-Agarose resin, prepared as described in Methods. Proteins were eluted with 400 μg/ml SIESDV peptide as described in Methods.
(C) Soluble lysates after heterologous expression of MBP; GST; single and tandem PDZbh domains; and MBP, GST, and MBP-GST fusion proteins containing zero, one or two N-terminal, C-terminal or internal PDZbh domains.
(D) Peptide eluates from purification of the proteins shown in (C) on PDZ2-HaloTag-HaloLink domain resin eluted with 400 μg/ml SIESDV peptide, as described in Methods.
PDZ domain beta-hairpin (PDZbh) as an affinity tag
Because of its small, modular structure, high solubility, high expression level in E. coli, and its affinity for the PDZ2 Domain Affinity Resin (described under “Purification of PDZbh from nNOS”), we reasoned that the PDZbh domain from nNOS might make an excellent N-terminal, C-terminal or internal PDZ Domain Affinity Tag. We also reasoned that two tandem PDZbh tags might have higher avidity for PDZ Domain Affinity Resins than single PDZbh tags. We tested these ideas in the experiment shown in (Fig. 7C and D). Positioning of a tandem PDZbh tag at the carboxyl terminus of MBP produced a slightly higher yield of the fusion protein than a single PDZbh tag, when the construct was purified on a PDZ2 Affinity Resin. However, a single PDZbh tag performed better than a tandem PDZbh tag when the tags were placed at the N-terminus or internally between MBP and GST in the fusion protein. PDZbh tags placed at the C-terminus or internally produced better yields than the corresponding tags placed at the N-terminus (Fig. 7D).
Functional Analysis of Purified Fusion Proteins
Tagging of proteins for functional tests
CAT, LacZ and DasherGFP proteins were recombinantly tagged with C-terminal PDZ2 Domain Peptide Ligand Affinity Tags and purified on PDZ2-NHS-Agarose resin, as described in Methods. They were purified to >95% homogeneity and recovered at concentrations of 17, 7.7, and 7.8 μM, with yields of 18.8, 8.4, and 8.6% of the column capacity for CAT, LacZ, and DasherGFP, respectively (Fig. 8A). In these experiments, the amount of POI in the bacterial lysate loaded onto the column greatly exceeded the column capacity.
Figure 8. Purification of Proteins Using PDZ Domain and PDZ Peptide Ligand Affinity Tags.
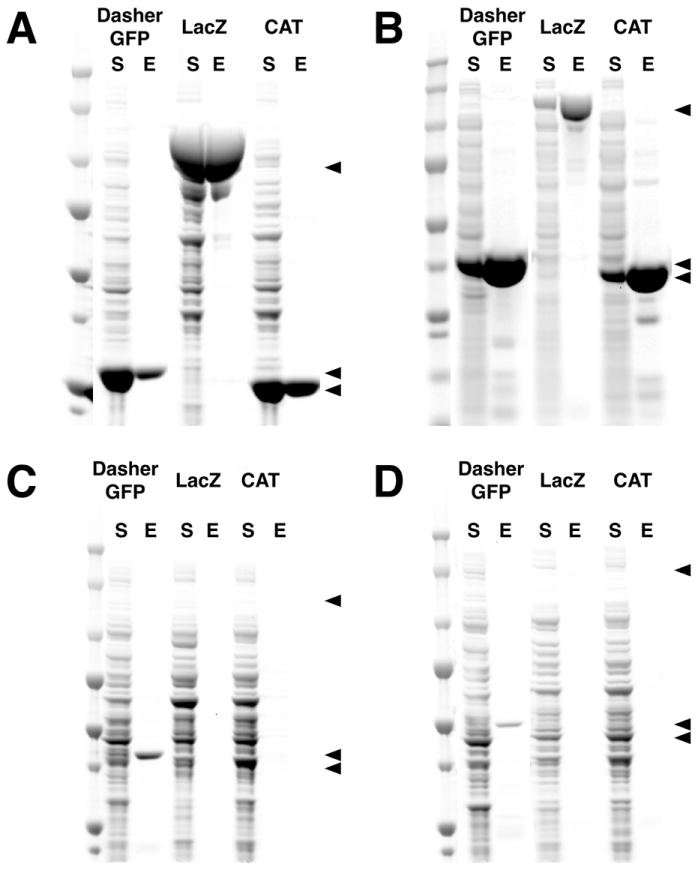
DasherGFP, LacZ and CAT were heterologously expressed with PDZ2 Domain or PDZ2 Peptide Ligand Tags and purified with the cognate Affinity Resin. For each protein, the soluble fraction (S) from the heterologous expression and the peptide eluate (E) after the purification were run on a 4-12% SDS-PAGE gradient gel with Precision Plus Protein All Blue Standards and stained with Gel Code Blue. (A) DasherGFP, LacZ and CAT were tagged at the C-terminus with the PDZ2 Domain Peptide Ligand, purified on PDZ2 Domain-NHS-Agarose Affinity Resin, and eluted with 400 μg/ml SIESDV peptide. (B) The three proteins were tagged at the N-terminus with a PDZ2 Domain, purified on PDZ2 Domain Peptide Ligand-NHS-Agarose resin, and eluted with 400 μg/ml SIESDV peptide. (C) The three proteins were tagged at the N-terminus with a PDZbh Domain or (D) two tandem PDZbh Domains, purified on PDZ2 Domain-NHS-Agarose Affinity Resin, and eluted with 400 μg/ml SIESDV peptide.
In the reverse experiment, the three proteins were tagged at the N-terminus with PDZ2 Domain Affinity Tags and purified on PDZ2 Domain Peptide Ligand Affinity Resin. They were purified to >95% homogeneity at concentrations of 70, 5.8, and 82 μM, with yields of 66, 81, and 37% for CAT, LacZ, and DasherGFP, respectively (Fig. 8B). A similar amount of bacterial lysate was loaded onto each column for the purifications shown in Figure 8A and B; however, the PDZ2 Domain Ligand Resin produced a greater yield of pure protein at a higher concentration because the density of Peptide Ligand on the column was 30-fold higher than the density of the PDZ2 Domain Resin (~20 nmol/μl vs. 0.724 nmol/μl). The lower concentration of LacZ eluted from the PDZ Domain Ligand Resin and its higher yield, compared to the amount loaded, were due in part to a 13-fold reduction in the level of bacterial expression of LacZ when the PDZ Domain Ligand Affinity Tag was fused to the C-terminus rather than to the N-terminus. These results illustrate that the optimum combination of PDZ-related Affinity Tag and Affinity Column for a particular POI will depend on the effect of the Tag on the protein’s expression and functional activity, as well as on the capacity of the cognate Affinity Resin.
Finally, CAT, LacZ and DasherGFP proteins were tagged N-terminally with single or tandem PDZbh Domain Affinity Tags and purified on PDZ2-NHS-Agarose resin. DasherGFP was purified to >95% homogeneity with a yield of 16%, but LacZ and CAT were not recovered in detectable quantities (Fig. 8C and D). We hypothesize that the inability to purify LacZ and CAT when fused to PDZbh Domain Affinity Tags may be caused by occlusion of the β-Hairpin domain in the fusion protein.
Function of DasherGFP
Green fluorescent protein (GFP) is a 238 residue protein isolated from the Pacific Northwest jellyfish, Aequeorea victoria [56]. GFP transmutes blue chemiluminescence from a primary photoprotein (aequorin) into green fluorescence [57], utilizing a p-hydroxybenzylidene-imidazolidone chromophore derived from its S65, Y66 and G67 residues [58]. It has been used in the production of biosensors for monitoring intracellular pH [59-61], calcium concentration [62], redox potential [63, 64], membrane potential [65] and temperature [66]. Proper folding of GFP around the chromophore is necessary for fluorescence, as evidenced by the fact that synthetic p-hydroxybenzylidene-imidazolidone chromophores are devoid of fluorescence [67]. When fused to the C-terminus of a POI, productive folding of the downstream GFP and formation of the fluorescent chromophore have been shown to depend on the robustness of folding of the upstream protein [68].
DasherGFP is a 26.6 kDa, synthetic, non-aequorea fluorescent protein, developed by DNA2.0, with excitation and emission wavelengths of 505 and 525 nm, respectively (Fig. 9A). To verify that PDZ Affinity Tags do not inhibit the fluorescence emission of DasherGFP, we performed excitation and emission wavelength scans on each purified, PDZ Affinity Tagged DasherGFP construct (Fig. 9B-E). Purified DasherGFP fused to a C-terminal PDZ Domain Peptide Ligand Tag (Fig. 9B), an N-terminal PDZ Domain Tag (Fig. 9C), or a single N-terminal PDZbh Domain Tag (Fig. 9D) exhibited fluorescence excitation and emission spectra that are virtually identical to the unlabeled protein (Fig. 9A). In contrast, DasherGFP fused to an N-terminal tandem PDZbh Domain Tag (Fig. 9E) had greater overlap between the two spectra than the unlabeled protein with excitation and emission maxima both occuring at 510 nm. To estimate the percentage of folded and functional tagged DasherGFP in each purified sample, the fluorescence intensity at 520 nm of each tagged DasherGFP was compared to that of an equimolar amount of untagged DasherGFP (Table 3). The fusions with a C-terminal PDZ Domain Peptide Ligand, an N-terminal PDZ Domain, or a single N-terminal PDZbh Domain all exhibited relative fluorescence intensities of greater than 90%, indicating that they are almost entirely folded and functional. However, the fusion with tandem PDZbh Domain Tags was almost entirely misfolded (Table 3), again suggesting that the tandem PDZbh Domain Affinity Tag destabilized proper folding.
Figure 9. Functional Analysis of Purified DasherGFP Fusion Proteins.
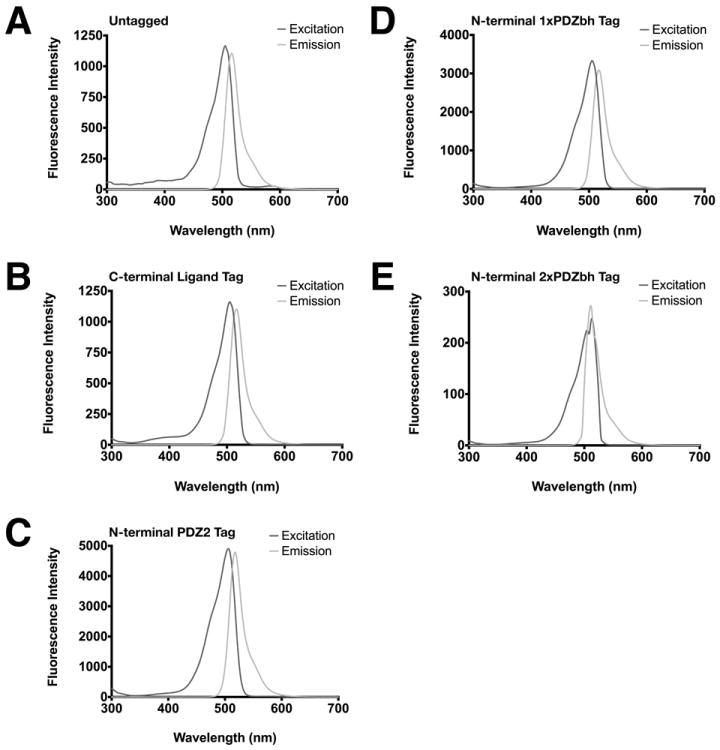
Fluorescence excitation and emission spectra of DasherGFP and DasherGFP fusion proteins were measured as described in Methods. (A) untagged; (B) fused to C-terminal PDZ Domain Peptide Ligand; (C) fused to N-terminal PDZ2 Domain; (D) fused to single PDZbh Domain; (E) fused to two tandem PDZbh Domains.
Table 3. Functional Analysis of DasherGFP and Purified DasherGFP Fusion Proteins.
Fluorescence emission at 520 nm, relative to untagged DasherGFP, of equimolar amounts of purified DasherGFP fused to a C-terminal PDZ2 Domain Peptide Ligand, an N-terminal PDZ2 Domain, and single or tandem PDZbh Domains.
| Affinity Tag | Relative Fluorescence at 520 nm (%) |
|---|---|
| Untagged DasherGFP | 100 |
| C-terminal PDZ Domain Peptide Ligand | 99.2 |
| N-terminal PDZ Domain | 97.5 |
| N-terminal 1x PDZ Domain+Beta Hairpin | 90.1 |
| N-terminal 2x PDZ Domain+Beta Hairpin | 0.3 |
Function of LacZ
β-Galactosidase (LacZ) is a 1024 residue protein isolated from E. coli [69, 70]. It catalyzes the cleavage of the bond between the anomeric carbon and glycosyl oxygen of a β-D-galactopyranoside [71]. In vivo LacZ catalyzes the cleavage of the disaccharide lactose to form glucose and galactose [72]. It is often used experimentally as a reporter of gene expression, of spontaneous or directed genetic changes in coding sequences, and of protein-protein interactions because of its activity in myriad cell lines [73-77], the availability of an array of substrates, inducers and inhibitors [77-79], its structural malleability [80-82] and the large dynamic range of its gene expression [83].
To verify that PDZ Affinity Tags do not inhibit the enzyme activity of LacZ, we assayed each tagged LacZ using ONPG as a substrate, as described in Methods (Fig. 10). The catalytic constants (kcat = Vmax/[E]) of LacZ fused to a C-terminal PDZ Domain Peptide Ligand (Fig. 10A), or an N-terminal PDZ Domain (Fig. 10B), were in good agreement with the published kcat for untagged LacZ under identical conditions (Table 4). The KM’s for ONPG of these two tagged versions of LacZ were also in the same range as those published for untagged LacZ (Table 4). When compared by ordinary one way ANOVA, the kcat (p ≥ 0.16) and KM (p ≥ 0.21) values of tagged LacZ were not significantly different from those of untagged LacZ. Thus, LacZ engineered to contain either of these affinity tags and purified by PDZ Affinity Chromatography is folded and functional. LacZ fused N-terminally to single or tandem PDZbh Domain Tags could not be purified by Affinity Chromatography and so was not assayed.
Figure 10. Functional Analysis of Purified LacZ Fusion Proteins.
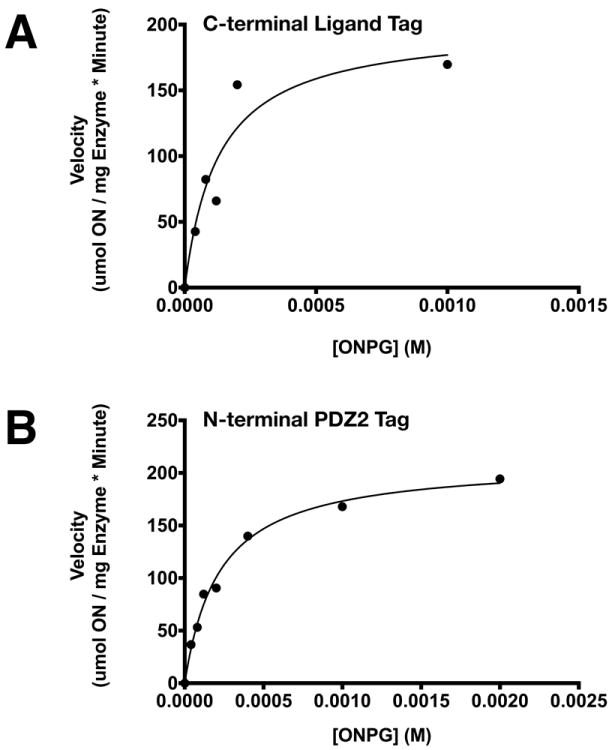
Michaelis-Menten plots for purified LacZ fused to (A) C-terminal PDZ2 domain peptide ligand; and (B) N-terminal PDZ2 domain. β-Galactosidase activity of LacZ was assayed by measuring hydrolysis of 0-2 mM ONPG for 10 minutes at 20 °C. Michaelis-Menten constants (Table 4), were calculated as described in Methods.
Table 4. Michaelis-Menten Constants of LacZ and Purified LacZ Fusion Proteins.
Comparison of kcat and KM values for LacZ and purified LacZ fused to a C-terminal PDZ2 Domain Peptide Ligand or an N-terminal PDZ2 Domain.
| Affinity Tag | kcat (sec-1) | KM (μM) |
|---|---|---|
| None | 345* | 161* |
| C-terminal PDZ Domain Peptide Ligand | 413±38 | 136±44 |
| N-terminal PDZ Domain | 390±10 | 227±21 |
Wallenfels K. and Malhotra P. (1962) Advanced Carbohydrate Chemistry: 239-298
Function of Chloramphenicol Acetyltransferase (CAT)
Chloramphenicol acetyltransferase (CAT) is a ~219 residue protein isolated from E. coli and S. aureus. It catalyzes the inactivation of the antibiotic chloramphenicol, by acylating chloramphenicol in the presence of acetyl-CoA to produce chloramphenicol-3-acetate and reduced CoA [84, 85]. It has been used extensively as an in vitro reporter of gene expression levels in eukaryotic cell lines because of its stability, absence of competing activities in eukaryotic cells, ease of use and sensitivity [86-88].
To verify that PDZ Affinity Tags do not inhibit the enzymatic activity of CAT, we measured activity of untagged CAT and each tagged construct using chloramphenicol and acetyl-CoA as substrates, as described in Methods (Fig. 11). Untagged CAT (Fig. 11A) and CAT fused to a C-terminal PDZ Domain Peptide Ligand (Fig. 11B), or an N-terminal PDZ Domain (Fig. 11C) were assayed with a fixed concentration of acetyl-CoA (500 μM) and 0-100 μM chloramphenicol. Their measured catalytic constants were in good agreement with the kcat’s of untagged CAT purchased from Sigma Aldrich (Fig. 11A; Table 5). The KM’s for chloramphenicol of these two tagged versions of CAT are similar to those measured for untagged CAT (Table 5) and published values [10 μM; 25]. When compared by ordinary one way ANOVA, the kcat (p ≥ 0.61) and KM (p ≥ 0.12) values of tagged CAT for chloramphenicol were not significantly different from those of untagged CAT.
Figure 11. Functional Analysis of Purified Chloramphenicol Acetyl Transferase (CAT) Fusion Proteins.
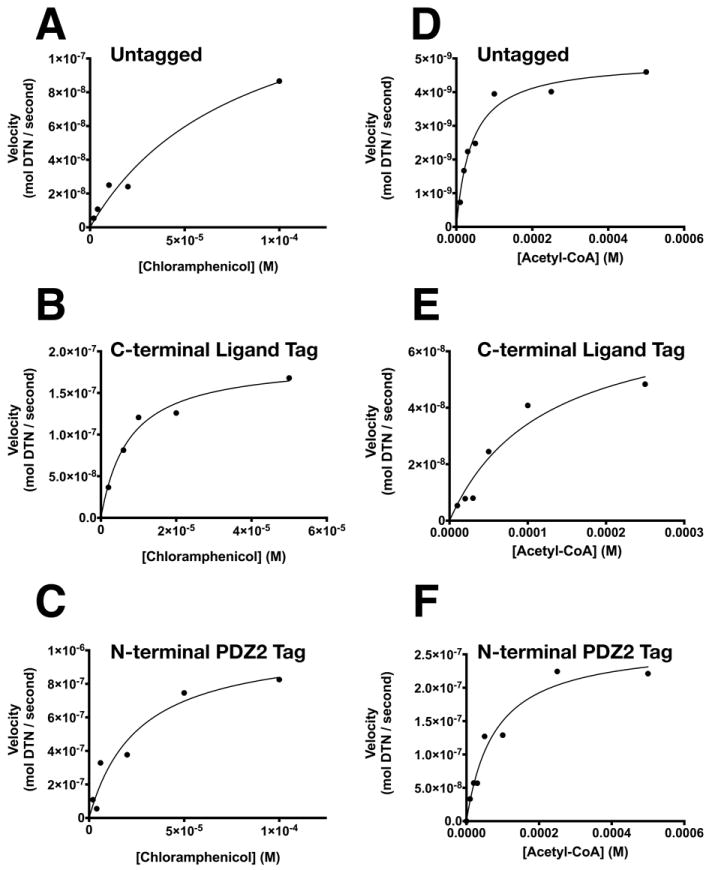
Michaelis-Menten plots for purified CAT: (A and D) untagged; (B and E) fused to a C-terminal PDZ2 domain peptide ligand; and (C and F) fused to an N-terminal PDZ2 domain. Transferase activity of purified CAT was assayed with (A-C) 500 μM Acetyl-CoA and 0-100 μM Chloramphenicol, or with (D-F) 100 μM Chloramphenicol and 0-500 μM Acetyl-CoA for 10 minutes at 37 °C. Michaelis-Menten constants (Table 5), were calculated as described in Methods.
Table 5. Michaelis-Menten constants of CAT and Purified CAT fusion proteins.
Comparison of kcat and KM values for CAT and purified CAT fused to a C-terminal PDZ2 Domain Peptide Ligand or an N-terminal PDZ2 Domain.
| Chloramphenicol** | Acetyl-CoA*** | |||
|---|---|---|---|---|
| Affinity Tag | kcat (sec-1) | KM (μM) | kcat (sec-1) | KM (μM) |
| None* | 0.43 ± 0.11 | 80 ± 40 | 0.085 ± 0.004 | 41 ± 7 |
| C-terminal PDZ Domain Peptide Ligand | 0.37 ± 0.03 | 8 ± 2 | 0.074 ± 0.02 | 120 ± 50 |
| N-terminal PDZ Domain | 0.37 ± 0.06 | 26 ± 10 | 0.093 ± 0.008 | 78 ± 20 |
CAT was purchased from Sigma Aldrich (Cat. No. C8413, St. Louis MO).
Chloramphenicol concentration ranged from 0-100 μM. Acetyl-CoA concentration was fixed at 500 μM.
Acetyl-CoA concentration ranged from 0-500 μM. Chloramphenicol concentration was fixed at 100 μM.
When the two tagged versions of CAT (Fig. 11E and F) were assayed with a fixed concentration of chloramphenciol (100 μM) and 0-500 μM acetyl-CoA, their catalytic constants were also in good agreement with the kcat’s of untagged CAT purchased from Sigma (Fig. 11D; Table 5) and published values [50 μM; 25]. The KM’s for acetyl-CoA of these two tagged versions of CAT were also similar to published values for untagged CAT (Table 5). When compared by ordinary one way ANOVA, the kcat (p ≥ 0.37) and KM (p ≥ 0.17) values of tagged CAT for acetyl-CoA were not significantly different from those of untagged CAT. Thus, CAT engineered to contain either of these affinity tags and purified by PDZ Domain Affinity Chromatography is folded and functional. As was the case for LacZ, CAT fused N-terminally to single or tandem PDZbh Domain Tags could not be purified by Affinity Chromatography and so were not assayed.
Conclusion
The PDZ Domain Affinity Chromatography system reported herein adds several additional tools to the toolbox of Affinity Tags and Affinity Resins available for protein purification. When compared to the His Tag system, the C-terminal PDZ Domain Peptide Ligand Affinity Tag and PDZ Domain Affinity Resin have several advantages: the affinity of the tag for the resin is generally higher (0.1 to 1 μM [13-15] versus 1-13 μM [89-93]); elution can be carried out with a solution of free peptide without the possibility of toxic metal ion (Zn2+, Co2+, Ni2+) contamination by leakage off the column. A potential disadvantage of the PDZ Domain Affinity Chromatography system is that, unlike the His Tag system, it cannot be used under denaturing conditions. When compared to the GST Tag system, the PDZ Domain Affinity System also has advantages: again, the affinity of the tag for the affinity resin is higher (0.1 to 1 μM [13-15] versus 40-170 μM [94-98]); it can be used in the presence of reducing agents; and the affinity tags are monomeric, whereas glutathione is dimeric or multimeric. The PDZ Domain-related Affinity Resins do not show detectable background binding of E. coli host cell proteins [89]. However, it is possible that endogenous proteins in eukaryotic cells that contain either PDZ domains or PDZ domain ligands will reduce the utility of PDZ-domain chromatography for purification of proteins from eukaryotic tissues or after expression in eukaryotic systems.
The potential to mix and match various related affinity tags and affinity resins adds flexibility to the PDZ Domain Affinity System. POIs lacking endogenous PDZ Domain Ligands can be engineered as fusion products with C-terminal PDZ Domain Ligand Affinity Tags and purified on a PDZ Domain Affinity Resin as outlined in Fig. 1. We suggest that a particularly useful combination will be tagging of POIs with the GluN2B ligand sequence (SSIESDV) and purification on a PDZ2 Domain Affinity Resin. The tagged POI can be eluted with a solution of free GluN2B Peptide Ligand (SIESDV). Other alternatives include engineering N-terminal, C-terminal, or internal PDZ Domain Tags into the POI followed by purification on PDZ Domain Ligand Affinity Resin as outlined in Fig. 2. We suggest tagging POIs with PDZ2 from PSD-95 and purifying the resulting proteins using a GluN2B Ligand-Resin (GAGSSIESDV), which can be synthesized at high capacity. Elution can be carried out with a solution of free GluN2B Ligand (SIESDV). This combination will be particularly useful for applications requiring purification of several milligrams of protein. The best choice of resin and tag pairs will ultimately depend on the expression level, stability, and solubility of the POI when fused to either of the two potential classes of tags. We have shown that CAT, LacZ and DasherGFP fused to either an N-terminal PDZ Domain Tag or to a C-terminal PDZ Domain Peptide Ligand Affinity Tag could all be purified in fully active form on the cognate Affinity Resin.
Supplementary Material
Fractions from attempted expression and purification of proteins on commercially available Affinity Resins and Precision Plus Protein All Blue Standards were separated on 4-12% gradient SDS-PAGE gels and stained with Gel Code Blue.
(A) The GluN2B Tail expressed without a tag was insoluble after expression in E. coli. T, total bacterial lysate; S, soluble bacterial protein; and I, insoluble bacterial protein pelleted by centrifugation.
(B) The GluN2B Tail expressed as an N-terminal MBP fusion protein (MBP-GluN2B Tail) was soluble and could be purified with amylose affinity resin (E8021; New England Biolabs, Ipswitch MA) and eluted with 10 mM maltose. The product was approximately 8% pure and was recovered at a concentration of 0.1 μM with a yield of 6%. T, total protein; S, soluble protein; and E, eluate.
(C) R-synGAP was purified with Talon Cobalt-CMA affinity resin (Cat. No. 635503; Clontech Laboratories, Inc., Mountain View CA) and eluted with 10 mM EDTA. The product was approximately 25% pure and was recovered at a concentration of 2 μM with a yield of 75%. T, total protein; S, soluble protein; and E, eluate.
Heterologous expression and purification of MBP fused to a truncation library of the C-terminal ten residues of the GluN2B Tail. All samples and Precision Plus Protein All Blue Standards were fractionated on a 4-12% gradient SDS-PAGE gel and stained with Gel Code Blue. (A) Clarified lysate after bacterial expression. (B) Eluates from purification of MBP-GluN2B Tail C-terminal truncations on PDZ2-HaloTag-HaloLink Resin, eluted with 400 μg/ml SIESDV peptide. The construct for expression of MBP-ESDV was resequenced and is correct. This fusion protein runs anomalously on SDS-PAGE.
Heterologous expression and purification of thioredoxin (THX) fused to a truncation library of the C-terminal ten residues of the GluN2B Tail. All samples and Precision Plus Protein All Blue Standards were fractionated on a 4-12% gradient SDS-PAGE gel and stained with Gel Code Blue. (A) Clarified lysate after bacterial expression. (B) Eluate from purification of THX-GluN2B Tail C-terminal truncations on PDZ2-HaloTag-HaloLink Resin, eluted with 400 μg/ml SIESDV peptide.
Highlights.
A chromatography method for purifying proteins containing PDZ domain-binding ligands
PDZ domain-binding ligands can be naturally occurring or introduced by cloning
Matrices consist of PDZ domains or their ligands, with free ligand used for elution
We purified heterologously expressed neuronal proteins containing PDZ domain ligands
Using this method enzymes and fluorescent proteins were purified in their active form
Acknowledgments
This work was supported by grants from the Gordon and Betty Moore Foundation, the Hicks Foundation for Alzheimer’s Research, the Allen and Lenabelle Davis Foundation, and from National Institutes of Health Grant MH095095 to MBK. WGW IV was supported by the National Science Foundation Graduate Research Fellowship under Grant No. 2006019582 and the National Institutes of Health under Grant No. NIH/NRSA 5 T32 GM07616.
We thank Kathryn M. Ivanetich for her careful editing and review of the manuscript, Jost Vielmetter and Mark Welch for technical discussions and Leslie Schenker for technical assistance.
Footnotes
Author Contributions: WGWIV conceived, designed, and carried out experiments; analyzed and interpreted data; and wrote the paper. MBK interpreted data and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Kornau H-C, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 2.Kornau H-C, Seeburg PH, Kennedy MB. Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol. 1997;7:368–373. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- 3.Hung AY, Sheng M. PDZ domains: structural modules for protein complex assembly. J Biol Chem. 2002;277:5699–5702. doi: 10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- 4.Cho K-O, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 5.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 6.Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell. 1996;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- 7.Ivarsson Y, Wawrzyniak AM, Wuytens G, Kosloff M, Vermeiren E, Raport M, Zimmermann P. Cooperative phosphoinositide and peptide binding by PSD-95/discs large/ZO-1 (PDZ) domain of polychaetoid, Drosophila zonulin. J Biol Chem. 2011;286:44669–44678. doi: 10.1074/jbc.M111.285734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric-oxide synthase with the postsynaptic density protein PSD-95 and a-1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 9.Niethammer M, Valtschanoff JG, Kapoor TM, Allison DW, Weinberg RJ, Craig AM, Sheng M. CRIPT, a novel postsynaptic protein that binds to the third PDZ domain of PSD-95/SAP90. Neuron. 1998;20:693–707. doi: 10.1016/s0896-6273(00)81009-0. [DOI] [PubMed] [Google Scholar]
- 10.Marfatia SM, Morais Cabral JH, Lin L, Hough C, Bryant PJ, Stolz L, Chishti AH. Modular organization of the PDZ domains in the human discs-large protein suggests a mechanism for coupling PDZ domain-binding proteins to ATP and the membrane cytoskeleton. J Cell Biol. 1996;135:753–766. doi: 10.1083/jcb.135.3.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC. Sap102, a novel postsynaptic protein that interacts with NMDA receptor complexes in-vivo. Neuron. 1996;17:255–265. doi: 10.1016/s0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- 12.Harris BZ, Hillier BJ, Lim WA. Energetic determinants of internal motif recognition by PDZ domains. Biochemistry. 2001;40:5921–5930. doi: 10.1021/bi010142l. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Piserchio A, Mierke DF. Structural characterization of the intermolecular interactions of synapse-associated protein-97 with the NR2B subunit of N-methyl-D-aspartate receptors. The Journal of biological chemistry. 2005;280:26992–26996. doi: 10.1074/jbc.M503555200. [DOI] [PubMed] [Google Scholar]
- 14.Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–21711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- 15.Lim IA, Merrill MA, Chen Y, Hell JW. Disruption of the NMDA receptor-PSD-95 interaction in hippocampal neurons with no obvious physiological short-term effect. Neuropharmacology. 2003;45:738–754. doi: 10.1016/s0028-3908(03)00276-4. [DOI] [PubMed] [Google Scholar]
- 16.Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron. 1993;11:531–540. doi: 10.1016/0896-6273(93)90157-m. [DOI] [PubMed] [Google Scholar]
- 17.Hillier BJ, Christopherson KS, Prehoda KE, Bredt DS, Lim WA. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science. 1999;284:812–815. [PubMed] [Google Scholar]
- 18.Tochio H, Zhang Q, Mandal P, Li M, Zhang M. Solution structure of the extended neuronal nitric oxide synthase PDZ domain complexed with an associated peptide. Nat Struct Biol. 1999;6:417–421. doi: 10.1038/8216. see comments. [DOI] [PubMed] [Google Scholar]
- 19.Kimple ME, Sondek J. Affinity tag for protein purification and detection based on the disulfide-linked complex of InaD and NorpA. Biotechniques. 2002;33:578, 580. doi: 10.2144/02333rr01. 584-578 passim. [DOI] [PubMed] [Google Scholar]
- 20.Klock HE, Koesema EJ, Knuth MW, Lesley SA. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins. 2008;71:982–994. doi: 10.1002/prot.21786. [DOI] [PubMed] [Google Scholar]
- 21.Klock HE, Lesley SA. The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol Biol. 2009;498:91–103. doi: 10.1007/978-1-59745-196-3_6. [DOI] [PubMed] [Google Scholar]
- 22.Wallenfels K. Beta-galactosidase. Meth Enzymol. 1962;5:212–218. [Google Scholar]
- 23.Wallenfels K, Lehmann J, Malhotra OP. Research on lactose-splitting enzymes. VII The specificity of beta-galactosidase from E. coli ML 309. Biochem Z. 1960;333:209–225. [PubMed] [Google Scholar]
- 24.Craven GR, Steers E, Jr, Anfinsen CB. Purification, Composition, and Molecular Weight of the Beta-Galactosidase of Escherichia Coli K12. J Biol Chem. 1965;240:2468–2477. [PubMed] [Google Scholar]
- 25.Shaw WV. Chloramphenicol acetyltransferase from chloramphenicol-resistant bacteria. Methods Enzymol. 1975;43:737–755. doi: 10.1016/0076-6879(75)43141-x. [DOI] [PubMed] [Google Scholar]
- 26.Winshell E, Shaw WV. Kinetics of induction and purification of chloramphenicol acetyltransferase from chloramphenicol-resistant Staphylococcus aureus. J Bacteriol. 1969;98:1248–1257. doi: 10.1128/jb.98.3.1248-1257.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 28.Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis. Methods Mol Biol. 2007;356:195–208. doi: 10.1385/1-59745-217-3:195. [DOI] [PubMed] [Google Scholar]
- 29.Hata T, Nakayama M. Rapid single-tube method for small-scale affinity purification of polyclonal antibodies using HaloTag Technology. J Biochem Biophys Methods. 2007;70:679–682. doi: 10.1016/j.jbbm.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- 32.Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunt CA, Schenker LJ, Kennedy MB. PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J Neurosci. 1996;16:1380–1388. doi: 10.1523/JNEUROSCI.16-04-01380.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 35.Seeburg PH. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- 36.Mayer ML, Armstrong N. Structure and function of glutamate receptor ion channels. Annu Rev Physiol. 2004;66:161–181. doi: 10.1146/annurev.physiol.66.050802.084104. [DOI] [PubMed] [Google Scholar]
- 37.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 38.Mayer ML. Glutamate receptors at atomic resolution. Nature. 2006;440:456–462. doi: 10.1038/nature04709. [DOI] [PubMed] [Google Scholar]
- 39.Kohr G, Jensen V, Koester HJ, Mihaljevic AL, Utvik JK, Kvello A, Ottersen OP, Seeburg PH, Sprengel R, Hvalby O. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 2003;23:10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H-J, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM Kinase II. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- 41.Kim JH, Lee HK, Takamiya K, Huganir RL. The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci. 2003;23:1119–1124. doi: 10.1523/JNEUROSCI.23-04-01119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vazquez LE, Chen HJ, Sokolova I, Knuesel I, Kennedy MB. SynGAP regulates spine formation. J Neurosci. 2004;24:8862–8872. doi: 10.1523/JNEUROSCI.3213-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O’Carroll CM, Martin SJ, Morris RG, O’Dell TJ, Grant SG. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci. 2002;22:9721–9732. doi: 10.1523/JNEUROSCI.22-22-09721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McMahon AC, Barnett MW, O’Leary TS, Stoney PN, Collins MO, Papadia S, Choudhary JS, Komiyama NH, Grant SG, Hardingham GE, Wyllie DJ, Kind PC. SynGAP isoforms exert opposing effects on synaptic strength. Nat Commun. 2012;3:900. doi: 10.1038/ncomms1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM, Wang Z, Carmant L, Mottron L, Beauchamp MH, Rouleau GA, Michaud JL. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry. 2011;69:898–901. doi: 10.1016/j.biopsych.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 46.Oh JS, Manzerra P, Kennedy MB. Regulation of the neuron-specific Ras GTPase activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:17980–17988. doi: 10.1074/jbc.M314109200. [DOI] [PubMed] [Google Scholar]
- 47.Pena V, Hothorn M, Eberth A, Kaschau N, Parret A, Gremer L, Bonneau F, Ahmadian MR, Scheffzek K. The C2 domain of SynGAP is essential for stimulation of the Rap GTPase reaction. EMBO Rep. 2008;9:350–355. doi: 10.1038/embor.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Passafaro M, Sala C, Niethammer M, Sheng M. Microtubule binding by CRIPT and its potential role in the synaptic clustering of PSD-95. Nat Neurosci. 1999;2:1063–1069. doi: 10.1038/15990. [DOI] [PubMed] [Google Scholar]
- 49.Brenman JE, Topinka JR, Cooper EC, McGee AW, Rosen J, Milroy T, Ralston HJ, Bredt DS. Localization of postsynaptic density-93 to dendritic microtubules and interaction with microtubule-associated protein 1A. J Neurosci. 1998;18:8805–8813. doi: 10.1523/JNEUROSCI.18-21-08805.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brenman JE, Bredt DS. Synaptic signaling by nitric oxide. Curr Opin Neurobiol. 1997;7:374–378. doi: 10.1016/s0959-4388(97)80065-7. [DOI] [PubMed] [Google Scholar]
- 52.Bredt DS, Snyder SH. Nitric oxide, a novel neuronal messenger. Neuron. 1992;8:3–11. doi: 10.1016/0896-6273(92)90104-l. [DOI] [PubMed] [Google Scholar]
- 53.Firestein BL, Brenman JE, Aoki C, Sanchez-Perez AM, El-Husseini AE, Bredt DS. Cypin: a cytosolic regulator of PSD-95 postsynaptic targeting. Neuron. 1999;24:659–672. doi: 10.1016/s0896-6273(00)81120-4. [DOI] [PubMed] [Google Scholar]
- 54.Charych EI, Akum BF, Goldberg JS, Jornsten RJ, Rongo C, Zheng JQ, Firestein BL. Activity-independent regulation of dendrite patterning by postsynaptic density protein PSD-95. J Neurosci. 2006;26:10164–10176. doi: 10.1523/JNEUROSCI.2379-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akum BF, Chen M, Gunderson SI, Riefler GM, Scerri-Hansen MM, Firestein BL. Cypin regulates dendrite patterning in hippocampal neurons by promoting microtubule assembly. Nat Neurosci. 2004;7:145–152. doi: 10.1038/nn1179. [DOI] [PubMed] [Google Scholar]
- 56.Morise H, Shimomura O, Johnson FH, Winant J. Intermolecular energy transfer in the bioluminescent system of Aequorea. Biochemistry. 1974;13:2656–2662. doi: 10.1021/bi00709a028. [DOI] [PubMed] [Google Scholar]
- 57.Prendergast FG, Mann KG. Chemical and physical properties of aequorin and the green fluorescent protein isolated from Aequorea forskalea. Biochemistry. 1978;17:3448–3453. doi: 10.1021/bi00610a004. [DOI] [PubMed] [Google Scholar]
- 58.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 59.Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 60.Hanson GT, McAnaney TB, Park ES, Rendell ME, Yarbrough DK, Chu S, Xi L, Boxer SG, Montrose MH, Remington SJ. Green fluorescent protein variants as ratiometric dual emission pH sensors. 1. Structural characterization and preliminary application. Biochemistry. 2002;41:15477–15488. doi: 10.1021/bi026609p. [DOI] [PubMed] [Google Scholar]
- 61.McAnaney TB, Park ES, Hanson GT, Remington SJ, Boxer SG. Green fluorescent protein variants as ratiometric dual emission pH sensors. 2. Excited-state dynamics. Biochemistry. 2002;41:15489–15494. doi: 10.1021/bi026610o. [DOI] [PubMed] [Google Scholar]
- 62.Mank M, Santos AF, Direnberger S, Mrsic-Flogel TD, Hofer SB, Stein V, Hendel T, Reiff DF, Levelt C, Borst A, Bonhoeffer T, Hubener M, Griesbeck O. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat Methods. 2008;5:805–811. doi: 10.1038/nmeth.1243. [DOI] [PubMed] [Google Scholar]
- 63.Ostergaard H, Henriksen A, Hansen FG, Winther JR. Shedding light on disulfide bond formation: engineering a redox switch in green fluorescent protein. EMBO J. 2001;20:5853–5862. doi: 10.1093/emboj/20.21.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 65.Baker BJ, Mutoh H, Dimitrov D, Akemann W, Perron A, Iwamoto Y, Jin L, Cohen LB, Isacoff EY, Pieribone VA, Hughes T, Knopfel T. Genetically encoded fluorescent sensors of membrane potential. Brain Cell Biol. 2008;36:53–67. doi: 10.1007/s11068-008-9026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wong FH, Banks DS, Abu-Arish A, Fradin C. A molecular thermometer based on fluorescent protein blinking. J Am Chem Soc. 2007;129:10302–10303. doi: 10.1021/ja0715905. [DOI] [PubMed] [Google Scholar]
- 67.Follenius-Wund A, Bourotte M, Schmitt M, Iyice F, Lami H, Bourguignon JJ, Haiech J, Pigault C. Fluorescent derivatives of the GFP chromophore give a new insight into the GFP fluorescence process. Biophys J. 2003;85:1839–1850. doi: 10.1016/S0006-3495(03)74612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 69.Kalnins A, Otto K, Ruther U, Muller-Hill B. Sequence of the lacZ gene of Escherichia coli. EMBO J. 1983;2:593–597. doi: 10.1002/j.1460-2075.1983.tb01468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fowler AV, Zabin I. Amino acid sequence of beta-galactosidase. XI. Peptide ordering procedures and the complete sequence. J Biol Chem. 1978;253:5521–5525. [PubMed] [Google Scholar]
- 71.Huber RE, Gupta MN, Khare SK. The active site and mechanism of the beta-galactosidase from Escherichia coli. Int J Biochem. 1994;26:309–318. doi: 10.1016/0020-711x(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 72.Huber RE, Kurz G, Wallenfels K. A quantitation of the factors which affect the hydrolase and transgalactosylase activities of beta-galactosidase (E. coli) on lactose. Biochemistry. 1976;15:1994–2001. doi: 10.1021/bi00654a029. [DOI] [PubMed] [Google Scholar]
- 73.Guarente L, Ptashne M. Fusion of Escherichia coli lacZ to the cytochrome c gene of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1981;78:2199–2203. doi: 10.1073/pnas.78.4.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guarente L. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 1983;101:181–191. doi: 10.1016/0076-6879(83)01013-7. [DOI] [PubMed] [Google Scholar]
- 75.Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- 76.Lis JT, Simon JA, Sutton CA. New heat shock puffs and beta-galactosidase activity resulting from transformation of Drosophila with an hsp70-lacZ hybrid gene. Cell. 1983;35:403–410. doi: 10.1016/0092-8674(83)90173-3. [DOI] [PubMed] [Google Scholar]
- 77.Hall CV, Jacob PE, Ringold GM, Lee F. Expression and regulation of Escherichia coli lacZ gene fusions in mammalian cells. J Mol Appl Genet. 1983;2:101–109. [PubMed] [Google Scholar]
- 78.Miller JH. A Short Course in Bacterial Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.: 1992. [Google Scholar]
- 79.Davies J, Jacob F. Genetic mapping of the regulator and operator genes of the lac operon. J Mol Biol. 1968;36:413–417. doi: 10.1016/0022-2836(68)90165-4. [DOI] [PubMed] [Google Scholar]
- 80.Ullmann A, Jacob F, Monod J. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the beta-galactosidase structural gene of Escherichia coli. J Mol Biol. 1967;24:339–343. doi: 10.1016/0022-2836(67)90341-5. [DOI] [PubMed] [Google Scholar]
- 81.Goldberg ME, Edelstein SJ. Sedimentation equilibrium of paucidisperse systems. Subunit structure of complemented beta-galactosidase. J Mol Biol. 1969;46:431–440. doi: 10.1016/0022-2836(69)90186-7. [DOI] [PubMed] [Google Scholar]
- 82.Muller-Hill B, Kania J. Lac repressor can be fused to beta-galactosidase. Nature. 1974;249:561–563. doi: 10.1038/249561a0. [DOI] [PubMed] [Google Scholar]
- 83.Fowler AV. High-level production of beta-galactosidase by Escherichia coli merodiploids. J Bacteriol. 1972;112:856–860. doi: 10.1128/jb.112.2.856-860.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shaw WV. The enzymatic acetylation of chloramphenicol by extracts of R factor-resistant Escherichia coli. J Biol Chem. 1967;242:687–693. [PubMed] [Google Scholar]
- 85.Suzuki Y, Okamoto S. The enzymatic acetylation of chloramphenicol by the multiple drug-resistant Escherichia coli carrying R factor. J Biol Chem. 1967;242:4722–4730. [PubMed] [Google Scholar]
- 86.Gorman CM, Moffat LF, Howard BH. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol Cell Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kain SR, Ganguly S. Overview of genetic reporter systems. Curr Protoc Mol Biol. 2001;Chapter 9(Unit9):6. doi: 10.1002/0471142727.mb0906s36. [DOI] [PubMed] [Google Scholar]
- 88.Thompson JF, Hayes LS, Lloyd DB. Modulation of firefly luciferase stability and impact on studies of gene regulation. Gene. 1991;103:171–177. doi: 10.1016/0378-1119(91)90270-l. [DOI] [PubMed] [Google Scholar]
- 89.Dorn IT, Neumaier KR, Tampé R. Molecular Recognition of Histidine-Tagged Molecules by Metal-Chelating Lipids Monitored by Fluorescence Energy Transfer and Correlation Spectroscopy§. Journal of the American Chemical Society. 1998;120:2753–2763. [Google Scholar]
- 90.Lata S, Reichel A, Brock R, Tampe R, Piehler J. High-affinity adaptors for switchable recognition of histidine-tagged proteins. Journal of the American Chemical Society. 2005;127:10205–10215. doi: 10.1021/ja050690c. [DOI] [PubMed] [Google Scholar]
- 91.Khan F, He M, Taussig MJ. Double-hexahistidine tag with high-affinity binding for protein immobilization, purification, and detection on ni-nitrilotriacetic acid surfaces. Analytical chemistry. 2006;78:3072–3079. doi: 10.1021/ac060184l. [DOI] [PubMed] [Google Scholar]
- 92.Nieba L, Nieba-Axmann SE, Persson A, Hamalainen M, Edebratt F, Hansson A, Lidholm J, Magnusson K, Karlsson AF, Pluckthun A. BIACORE analysis of histidine-tagged proteins using a chelating NTA sensor chip. Analytical biochemistry. 1997;252:217–228. doi: 10.1006/abio.1997.2326. [DOI] [PubMed] [Google Scholar]
- 93.Guignet EG, Hovius R, Vogel H. Reversible site-selective labeling of membrane proteins in live cells. Nature biotechnology. 2004;22:440–444. doi: 10.1038/nbt954. [DOI] [PubMed] [Google Scholar]
- 94.Fabrini R, De Luca A, Stella L, Mei G, Orioni B, Ciccone S, Federici G, Lo Bello M, Ricci G. Monomer-dimer equilibrium in glutathione transferases: a critical reexamination. Biochemistry. 2009;48:10473–10482. doi: 10.1021/bi901238t. [DOI] [PubMed] [Google Scholar]
- 95.Ivanetich KM, Goold RD. A rapid equilibrium random sequential bi-bi mechanism for human placental glutathione S-transferase. Biochimica et biophysica acta. 1989;998:7–13. doi: 10.1016/0167-4838(89)90111-8. [DOI] [PubMed] [Google Scholar]
- 96.Ivanetich KM, Goold RD, Sikakana CN. Explanation of the non-hyperbolic kinetics of the glutathione S-transferases by the simplest steady-state random sequential Bi Bi mechanism. Biochemical pharmacology. 1990;39:1999–2004. doi: 10.1016/0006-2952(90)90621-q. [DOI] [PubMed] [Google Scholar]
- 97.Ivanetich KM, Thumser AE, Harrison GG. Halothane: inhibition and activation of rat hepatic glutathione S-transferases. Biochemical pharmacology. 1988;37:1903–1908. doi: 10.1016/0006-2952(88)90535-7. [DOI] [PubMed] [Google Scholar]
- 98.Schramm VL, McCluskey R, Emig FA, Litwack G. Kinetic studies and active site-binding properties of glutathione S-transferase using spin-labeled glutathione, a product analogue. The Journal of biological chemistry. 1984;259:714–722. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fractions from attempted expression and purification of proteins on commercially available Affinity Resins and Precision Plus Protein All Blue Standards were separated on 4-12% gradient SDS-PAGE gels and stained with Gel Code Blue.
(A) The GluN2B Tail expressed without a tag was insoluble after expression in E. coli. T, total bacterial lysate; S, soluble bacterial protein; and I, insoluble bacterial protein pelleted by centrifugation.
(B) The GluN2B Tail expressed as an N-terminal MBP fusion protein (MBP-GluN2B Tail) was soluble and could be purified with amylose affinity resin (E8021; New England Biolabs, Ipswitch MA) and eluted with 10 mM maltose. The product was approximately 8% pure and was recovered at a concentration of 0.1 μM with a yield of 6%. T, total protein; S, soluble protein; and E, eluate.
(C) R-synGAP was purified with Talon Cobalt-CMA affinity resin (Cat. No. 635503; Clontech Laboratories, Inc., Mountain View CA) and eluted with 10 mM EDTA. The product was approximately 25% pure and was recovered at a concentration of 2 μM with a yield of 75%. T, total protein; S, soluble protein; and E, eluate.
Heterologous expression and purification of MBP fused to a truncation library of the C-terminal ten residues of the GluN2B Tail. All samples and Precision Plus Protein All Blue Standards were fractionated on a 4-12% gradient SDS-PAGE gel and stained with Gel Code Blue. (A) Clarified lysate after bacterial expression. (B) Eluates from purification of MBP-GluN2B Tail C-terminal truncations on PDZ2-HaloTag-HaloLink Resin, eluted with 400 μg/ml SIESDV peptide. The construct for expression of MBP-ESDV was resequenced and is correct. This fusion protein runs anomalously on SDS-PAGE.
Heterologous expression and purification of thioredoxin (THX) fused to a truncation library of the C-terminal ten residues of the GluN2B Tail. All samples and Precision Plus Protein All Blue Standards were fractionated on a 4-12% gradient SDS-PAGE gel and stained with Gel Code Blue. (A) Clarified lysate after bacterial expression. (B) Eluate from purification of THX-GluN2B Tail C-terminal truncations on PDZ2-HaloTag-HaloLink Resin, eluted with 400 μg/ml SIESDV peptide.