

Regulatory RNAs often contain long single-stranded RNA (ssRNA) regions that are typically found linked to helices and hairpins in what are sometimes referred to as ssRNA–helix junctions. This study uses NMR spectroscopy to study the dynamic properties of a 12-nucleotide (nt) ssRNA tail derived from the prequeuosine riboswitch linked to the 3′ end of a 48-nt hairpin. Despite substantial internal motions, the first two residues in the ssRNA tail stack onto the adjacent helix and assume an ordered conformation, and the ssRNA adopts, on average, an A-form helical conformation that is coaxial with the helix. Our results reveal a surprising degree of structural and dynamic complexity at the ssRNA–helix junction, which involves a fine balance between order and disorder that may facilitate efficient pseudoknot formation on ligand recognition.
Keywords: RNA dynamics, prequeuosine riboswitch, residual dipolar couplings, spin relaxation, ligand recognition
Abstract
Many regulatory RNAs contain long single strands (ssRNA) that adjoin secondary structural elements. Here, we use NMR spectroscopy to study the dynamic properties of a 12-nucleotide (nt) ssRNA tail derived from the prequeuosine riboswitch linked to the 3′ end of a 48-nt hairpin. Analysis of chemical shifts, NOE connectivity, 13C spin relaxation, and residual dipolar coupling data suggests that the first two residues (A25 and U26) in the ssRNA tail stack onto the adjacent helix and assume an ordered conformation. The following U26-A27 step marks the beginning of an A6-tract and forms an acute pivot point for substantial motions within the tail, which increase toward the terminal end. Despite substantial internal motions, the ssRNA tail adopts, on average, an A-form helical conformation that is coaxial with the helix. Our results reveal a surprising degree of structural and dynamic complexity at the ssRNA–helix junction, which involves a fine balance between order and disorder that may facilitate efficient pseudoknot formation on ligand recognition.
INTRODUCTION
Single-stranded RNAs (ssRNAs) are essential elements of RNA architecture and serve a wide variety of functions. They can act as spacers between structured domains (Lodeiro et al. 2009; Watts et al. 2009), provide binding sites for protein or RNA recognition (Auweter et al. 2006), act as checkpoints in RNA maturation (Spitzfaden et al. 2000), serve as signaling elements that can be sequestered into helices to generate switching behavior (Schwalbe et al. 2007), and form active sites to perform catalysis (Shi et al. 2012) and are key components of structured motifs such as pseudoknots (Zhang et al. 2011). Secondary structure analysis of RNA genomes and large structured RNAs reveals a pattern of adenine-enriched single-stranded regions (Gutell et al. 1985; Pollom et al. 2013).
ssRNAs are frequently considered to be unstructured despite considerable evidence to the contrary in certain sequences based on low-resolution structure characterization techniques such as circular dichroism (CD) and ultraviolet/visible (UV/Vis) spectroscopy (Dewey and Turner 1979; Freier et al. 1981). Recently we showed using a combination of NMR spectroscopy and replica exchange molecular dynamics (REMD) simulations that the adenine-rich, 12-nucleotide (nt) ssRNA in the Bacillus subtilis prequeuosine riboswitch adopts, on average, an A-form-like conformation with an ordered 6-nt adenine core and gradually increasing flexibility toward the terminal ends (Eichhorn et al. 2012a). Together with CD and UV/Vis melting experiments, these data indicated that the ssRNA is in equilibrium between an ordered A-form-like helical conformation and a highly disordered partially melted state. Similar observations have since been made on polycytosine ssRNA sequences using NMR, UV melting, and MD simulations, showing that ssRNA order is not limited to polyadenine sequences (Tubbs et al. 2013).
In RNA, ssRNA regions typically are found linked to helices and hairpins in what are sometimes referred to as ssRNA–helix junctions. Studies of ssRNA–helix junctions have largely focused on how ssRNA nucleotides that overhang the 3′ or 5′ termini stabilize the adjacent helix, ignoring the conformation and dynamics of the ssRNA residues themselves (O'Toole et al. 2005, 2006). Recently, Herschlag and coworkers studied the sequence dependence of an ssRNA adjoining the group I ribozyme to a duplex. The investigators found that for a three-adenine ssRNA junction, the adjoined duplex exhibited limited motions; however, a three-uridine junction greatly increased the duplex motions, nearly doubling the effective motional amplitude (Shi et al. 2012; Nguyen et al. 2013). While this study demonstrates the role of RNA sequence on interhelical dynamics and flexibility, little is known about the dynamic properties of ssRNA–helix junctions at the atomic level, even though they are often important sites for conformational changes and adaptation, particularly for systems containing pseudoknots (Kim et al. 2008; Cao and Chen 2009; Cash et al. 2013). The dynamic properties of ssRNA–helix junctions are of particular interest in the transcription-regulating prequeuosine riboswitch aptamer because the ssRNA tail must fold back onto the helix to form a pseudoknot to allow efficient cotranscriptional binding to ligand (Kang et al. 2009; Klein et al. 2009; Spitale et al. 2009; Rieder et al. 2010; Suddala et al. 2013). Our previous studies on the isolated 12-nt ssRNA indicated a degree of order within the ssRNA that may facilitate rapid docking into the minor groove of the adjacent helix in the presence of ligand (Eichhorn et al. 2012a); however, these studies neglected the effect of the ssRNA–helix junction.
Here, we examine the effects of local motions imparted by joining the ssRNA to a 48-nt hairpin—as occurs in biological RNAs—and also assess the dynamics of the ssRNA relative to the hairpin. The ssRNA–helix construct also allowed us to obtain more reliable estimates for absolute motional amplitudes within the ssRNA tail by decoupling internal and overall motions. Such a domain-elongation approach has been successfully used in the past to characterize motions in helix–junction–helix (HJH) motifs (Leeper and Varani 2005; Staple and Butcher 2005; Zhang et al. 2006, 2007, 2010; Getz et al. 2007b; Sun et al. 2007; Zhang and Al-Hashimi 2009).
RESULTS AND DISCUSSION
Construct design and resonance assignments
We previously reported the dynamic properties of an isolated 12-nt ssRNA, hereafter referred to as SS, derived from a B. subtilis prequeuosine riboswitch (Eichhorn et al. 2012a). We designed a ssRNA–helix junction construct variant containing the same 12-nt ssRNA connected to a GC-rich 22-bp hairpin capped with the thermodynamically stable cUUCGg tetraloop (E-SS) (Fig. 1A). The ssRNA is adjoined at the 3′ end of the helix, analogous to its position in the wild-type prequeuosine riboswitch. The GC-rich helix incorporates three isotopically (13C/15N) labeled A-U “reporter” base pairs that are used to obtain information regarding the dynamic properties of the helix. The construct was prepared using isotopically enriched 13C/15N A and U nucleotides and unlabeled G and C nucleotides, thus rendering the vast majority of resonances in the long helix “NMR invisible.” The helix was designed to be longer than that found in the prequeuosine riboswitch in order to help decouple internal and overall motions, thus facilitating quantitative analysis of NMR 13C spin relaxation and RDC data in describing both the local flexibility of the ssRNA and motions of the ssRNA relative to the helix across the ssRNA–helix junction (Zhang et al. 2006, 2007; Getz et al. 2007b; Sun et al. 2007).
FIGURE 1.
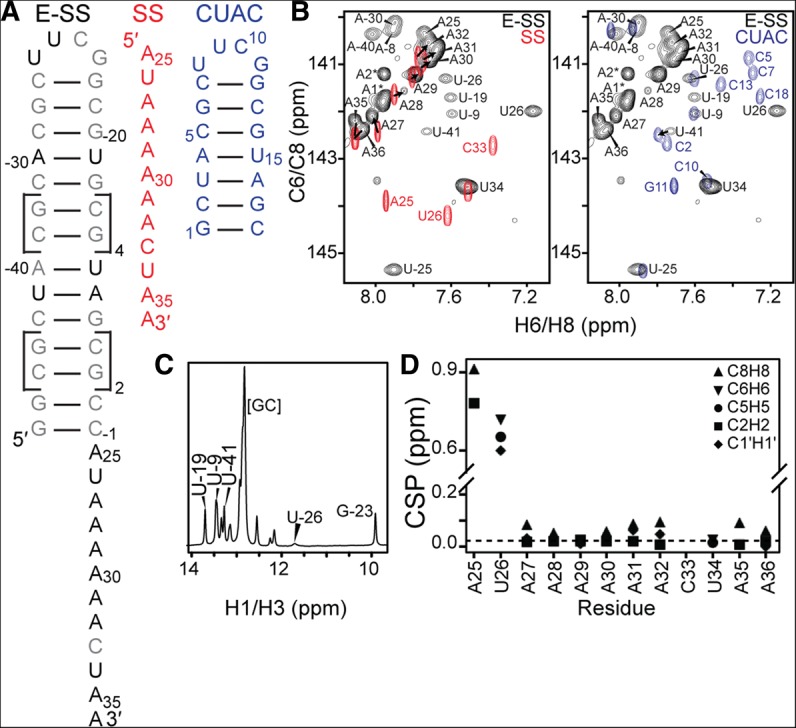
NMR chemical shift analysis of the ssRNA–helix junction. (A) Constructs used for NMR studies and resonance assignments. (B) Overlay of 2D 1H-13C HSQC spectra of SS and E-SS allows assignment of ssRNA residues (left), while overlay of E-SS and CUAC allows assignment of helical reporter residues (right). (C) 15N-filtered 1D 1H spectrum shows characteristic imino resonances for the helix and cUUCGg tetraloop. (D) Weighted average chemical shift perturbation (CSP) of ssRNA residues between the SS and E-SS constructs shows large perturbations for ssRNA–helix junction residues, while A27–A36 have minimal perturbations.
Analysis of 2D C-H and N-H HSQC NMR spectra confirmed that the E-SS construct folds into the predicted secondary structure (Fig. 1B,C). The imino 1H spectrum (Fig. 1C) revealed the expected number of base pairs within the helix. This, together with the observation of signature chemical shifts (CSs) for the cUUCGg tetraloop, confirmed that the hairpin adopts the expected helical conformation. In contrast, no imino resonances are observed for the ssRNA tail (Fig. 1C), indicating that it adopts a conformation lacking base-pairing interactions, as shown previously for the isolated ssRNA (Eichhorn et al. 2012a).
The resonances belonging to the reporter 13C/15N-labeled A and U residues embedded within the helix were assigned using a “divide and conquer” strategy similar to that used previously to assign elongated DNA helices (Nikolova and Al-Hashimi 2009). Specifically, we prepared an unlabeled NMR sample of a short (18-nt) helix containing the 5′-CUAC-3′ tetranucleotide step within the 22-bp helix, flanked on both 5′ and 3′ ends with two G-C base pairs and capped with a cUUCGg tetraloop (CUAC) (Fig. 1A,B). Resonances in this construct were easily assigned using standard 2D 1H-1H NOESY experiments and transferred to the E-SS construct based on excellent spectral overlap (Fig. 1B). As further confirmation of the assignments, resonances belonging to the 22-bp helix have the expected weakened resonance intensities compared with ssRNA residues, consistent with a slower overall tumbling rate (Fig. 1B).
With the exception of junction residues A25 and U26, we were able to transfer all of the sugar (C1′H1′) and nucleobase (C6H6, C8H8, C2H2, and C5H5) resonance assignments from SS to E-SS based on excellent spectral overlap (Fig. 1B). The resonance assignments in E-SS were independently confirmed using 2D HCN experiments. The resonances belonging to A25 and U26 in E-SS were perturbed relative to SS and were difficult to assign because of severe spectral overlap in 2D NOESY spectra of E-SS. However, these resonances could be assigned using 2D NOESY spectra acquired on a construct containing a shorter helix and ssRNA that minimizes resonance overlap (E-SSshort) (Fig. 2A). Although the A25 C1′H1′ resonance could be assigned in the E-SSshort spectrum, we were unable to unambiguously assign A25 C1′H1′ in E-SS; however, we did observe several resonances at the expected position that may reflect the conformational exchange and heterogeneity that disappears upon ligand binding (see below).
FIGURE 2.
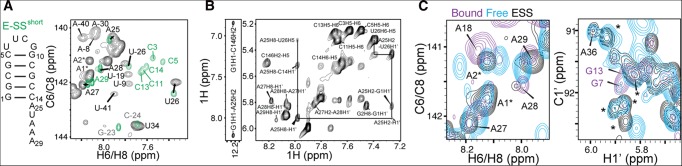
ssRNA–helix junction stacking interactions and comparison to a preQ1 riboswitch aptamer domain. (A) Comparison of 2D 1H-13C HSQC spectra (C6H6 and C8H8) for E-SS (black) and E-SSshort (green) allows assignment of ssRNA–helix junction residues. (B) 2D 1H-1H NOESY spectra of E-SSshort show NOE connectivities between the terminal G1-C14 base pair and ssRNA–helix junction, indicating ssRNA residues stack on the helix. (C) Comparison of 2D 1H-13C HSQC spectra (aromatic C6H6 and C8H8 and ribose C1′H1′) of E-SS (black) to Bsu preQ1-I riboswitch in the absence (cyan) and presence (purple) of preQ1 ligand. Additional adenine resonances that were unaccounted for are indicated using an asterisk.
Impact of helix on ssRNA conformation and dynamics from CS analysis
We examined how the addition of the hairpin affects the CSs observed for the SS tail. In Figure 1D, we show the weighted (1H and 13C) CS differences between E-SS and SS. Significant CS perturbations (>0.5 ppm) are observed for junction residues A25 and U26, indicating a change in their local electronic environment due to addition of the hairpin (Fig. 1D). The specific upfield perturbations in both the 1H and 13C CSs for base moieties (C5H5, C6H6, C8H8, and C2H2) are consistent with a more helical conformation. Although analysis of the E-SS NOESY spectra was complicated by severe resonance overlap, in E-SSshort we observe NOE connectivities between the terminal G1-C14 base pair and the junction ssRNA residue A25 (G1H1-A25H2, C14H1′-A25H8, and G1H1′-A25H2), as well as between A25 and U26 (A25H2-U26H1′, A26H8-U26H5) (Fig. 2B). This indicates that A25 and U26 stack onto the adjoined helix rather than forming flexible terminal-like residues. Prior studies have shown that ssRNA nucleotides that overhang the 3′ or 5′ termini stabilize the adjacent helix (O'Toole et al. 2005, 2006). Much smaller (<0.1 ppm) CS perturbations are observed at other residues in the ssRNA tail, including the adenine core (A30–A32), which also suggests a more helical conformation (Fig. 1B). Interestingly, these CS perturbations appear to depend on the length of the helix and/or ssRNA and are generally smaller in E-SSshort (Fig. 2A).
We note that in E-SS but not E-SSshort, we observe additional (2x C8H8, 2x C2H2, and 5x C1′H1′) resonances that need to be accounted for. These resonances are sharp and have high intensities, indicating that they belong to highly disordered residues. They are apparent in freshly made samples and do not change over time. These resonances are overlapped in the 1H dimension and could not be unambiguously assigned in 2D NOESY spectra (Fig. 1B). Furthermore, based on 2D HCN experiments, two C8H8, two C2H2, and two C1′H1′ of these resonances belong to two adenine residues. The remaining three C1′H1′ resonances are clustered near the A25 CS position in the isolated SS construct, indicating the structure may be experiencing slow conformational exchange on the NMR timescale at the ssRNA–helix junction. While all five C1′H1′ resonance positions are consistent with a terminal-like residue, the C8H8 and C2H2 resonances are similar to those observed for adenine residues within the A6-tract. Nucleobase C8H8 resonances for these adenine residues are shifted upfield with decreasing temperature, indicating increased stacking consistent with other adenine residues within the A6-tract; however, peak intensities do not appear to significantly change relative to other ssRNA resonances, suggesting the population of this alternative state is not temperature dependent. Interestingly, these adenine resonances are also observed in spectra of the 36-nt prequeuosine aptamer in the absence of ligand, but they disappear upon ligand binding (Fig. 2C). These resonances are therefore unlikely to arise from UV chemical damage to the RNA (Greenfeld et al. 2011; Kladwang et al. 2012) or from N + 1 products during in vitro RNA transcription (Milligan et al. 1987; Cazenave and Uhlenbeck 1994; Pleiss et al. 1998; Helm et al. 1999). Rather, these additional resonances could reflect an alternative conformation for adenine residues A25, A27, and A28 near the junction that is in slow exchange with the major state. Integration of adenine resonance volumes suggests that A27 and A28 could be experiencing the exchange due to reduced volumes relative to other adenine residues within the A6-tract (data not shown). Analysis of RDCs and 13C relaxation values measured for these resonances (Supplemental Tables S1, S2) suggests that these additional resonances belong to disordered residues, with near-zero RDCs and low R2/R1 values.
Picosecond–nanosecond motions from 13C spin relaxation
To examine the dynamic properties of E-SS at picosecond-to-nanosecond timescales, we measured 13C longitudinal (R1) and transverse (R2) spin relaxation data in E-SS for the nucleobase C2, C6, and C8 carbons as previously described (Eichhorn et al. 2012a). This allowed us to examine how the adjoined hairpin impacts local motions in the ssRNA tail, as well as obtain insights into motions of the ssRNA tail relative to the helix. Our previous 13C relaxation NMR studies of isolated SS made it difficult to obtain insights into the absolute amplitudes of internal motions owing to significant correlations between the internal and overall motions and lack of a frame of reference for characterizing overall motions (Zhang et al. 2006). In the E-SS construct, the adjoined helix helps decouple internal and overall motions and provides a reference for assessing overall motions, making it easier to measure the absolute level of motions in the ssRNA tail. This domain-elongation strategy has widely been applied in studies of RNA systems, but never for an ssRNA tail (Getz et al. 2007a; Bothe et al. 2011).
In Figure 3A we compare the R2/R1 values measured for each carbon site in the isolated SS construct with those measured when adjoining the helix. The R2/R1 values provide a measure of the extent of internal and overall motions occurring at nanosecond timescales. In the isolated SS, we observe a pattern in which the central adenine residues A29–A32 are the most ordered and have the highest R2/R1 values (Eichhorn et al. 2012a). As residues extend away from the polyadenine core, the levels of dynamics gradually increase, with maximum dynamics observed for the terminal residues A25 and A36 (Eichhorn et al. 2012a). Interestingly, the addition of the helix does not significantly affect the overall dynamics pattern in the ssRNA tail (Fig. 3A). However, there are two notable differences. First, while the dynamics of A27 are similar to that in the isolated SS, there is an abrupt decrease in dynamics at the junction residues A25 and U26, which have significantly elevated R2/R1 values that are comparable to those measured in the reporter A-U base pairs within the helix (Fig. 3A). This is consistent with CS and NOE data indicating stacking of these junction residues on the adjoined helix. Thus the pivot point for dynamics between the ssRNA tail and the helix is not the point of attachment (A25) but is rather the U26-A27 step, which is known to be highly flexible and which precedes the stable A6-tract in the ssRNA tail. Second, the R2/R1 values of A29–A32 in the A6-tract are significantly larger (about fourfold) in E-SS compared with SS. This may reflect comparatively greater stability within the ssRNA core due to the addition of the helix, or more likely, it reflects slower overall tumbling of E-SS compared with SS, which makes it possible to capture slower motions within the ssRNA tail. Independent support for the latter comes from the improved agreement with motional amplitudes derived from REMD simulations (see below).
FIGURE 3.
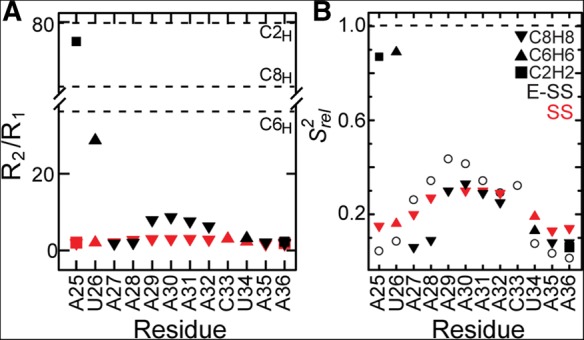
Dynamics of ssRNA–helix junction at picosecond–nanosecond timescales using carbon spin relaxation. Comparison of R2/R1 values (A) and relative spin relaxation order parameters  (B) for E-SS and SS. The SS
(B) for E-SS and SS. The SS 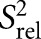 values are scaled relative to E-SS. Shown in open circles are the
values are scaled relative to E-SS. Shown in open circles are the 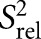 values computed for SS using REMD simulations (Eichhorn et al. 2012a). Dashed lines indicate average R2/R1 values for helical residues (C2H2, ∼80; C8H8, ∼60; C6H6, ∼40; A) or normalized S2 value (1; B).
values computed for SS using REMD simulations (Eichhorn et al. 2012a). Dashed lines indicate average R2/R1 values for helical residues (C2H2, ∼80; C8H8, ∼60; C6H6, ∼40; A) or normalized S2 value (1; B).
We also compared the 2R2 − R1 values for each carbon site, which is a good approximation for the order parameter S2, describing the amplitude of motions occurring at rates faster than the overall molecular tumbling, and which varies between zero and one for maximum and minimum motions (Fushman et al. 1999; Hansen and Al-Hashimi 2007). We computed relative S2
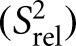 values by normalizing the 2R2 − R1 values for each carbon type (C2, C6, C8) relative to the largest value measured in the stable A-form helix (Fig. 3B; Hansen and Al-Hashimi 2007). Similar
values by normalizing the 2R2 − R1 values for each carbon type (C2, C6, C8) relative to the largest value measured in the stable A-form helix (Fig. 3B; Hansen and Al-Hashimi 2007). Similar 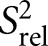 values of 0.3 are observed for the polyadenine tract in E-SS and SS, which agrees well with computed
values of 0.3 are observed for the polyadenine tract in E-SS and SS, which agrees well with computed 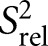 values from the previous REMD simulations (Eichhorn et al. 2012a). On the other hand, due to the slowing down of overall tumbling, larger motional amplitudes (lower
values from the previous REMD simulations (Eichhorn et al. 2012a). On the other hand, due to the slowing down of overall tumbling, larger motional amplitudes (lower 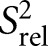 values) are observed for residues near the terminal ends, which are in better agreement with the REMD simulation (Fig. 3B, open circles). Many of these dynamics observations are mirrored independently in measured RDCs (see below).
values) are observed for residues near the terminal ends, which are in better agreement with the REMD simulation (Fig. 3B, open circles). Many of these dynamics observations are mirrored independently in measured RDCs (see below).
Global structure and sub-millisecond motions from RDCs
We measured RDCs (Tolman et al. 1995; Tjandra and Bax 1997) to gain further insights in the structural and dynamic behavior of the ssRNA–helix junction. RDCs depend on the orientation of a bond vector relative to an order tensor frame describing the average orientation of an aligned molecule relative to the applied magnetic field (Tolman et al. 1995; Tjandra and Bax 1997). They provide long-range structural information and are also sensitive to motions occurring over a broad range of timescales (picoseconds to milliseconds) (Tolman et al. 1997; Peti et al. 2002; Tolman and Ruan 2006; Getz et al. 2007a; Bothe et al. 2011; Eichhorn et al. 2012b).
We measured RDCs for base C5H5, C6H6, C8H8, and C2H2 and ribose C1′H1′ moieties by aligning E-SS in ∼8 mg/mL Pf1 phage (Fig. 4A; Clore et al. 1998; Hansen et al. 2000). Since the elongated helix is expected to be locally rigid and to partially dominate alignment with its long helical axis aligned on average nearly parallel to the magnetic field, nucleobases within the helix are expected to be aligned nearly perpendicular to the principal direction of order (Szz) describing the average direction of the molecule relative to the magnetic field (Zhang et al. 2007). Accordingly, the C-H RDCs measured in the nucleobases and sugars have the expected positive and negative values, respectively.
FIGURE 4.

RDC analysis of global structure and dynamics of the ssRNA–helix junction. (A) RDCs measured in E-SS. Dashed vertical lines separate the UUCG tetraloop, helical residues, and ssRNA residues. (B) Comparison of measured RDCs and back-calculated values for the 60-nt E-SS construct using the best-fit order tensor. The correlation constant (R2) and root mean square deviation (RMSD) are given at the top.
Toward the ssRNA tail, the residue-specific trends in the RDCs are similar to those observed with 13C spin relaxation. The magnitude of RDCs measured for junction residues A25 and U26 approaches those of the helical residues, consistent with stacking of these residues on the helix and with localization of the pivot points for motions between the ssRNA tail and helix not at the point of attachment (A25) but rather at the U26-A27 step, consistent with the 13C spin relaxation data. Once again, we observe an abrupt reduction in the magnitude of RDCs measured in A27, and the RDC values subsequently increase toward the core of the ssRNA, reaching a near maximum value at A30, before beginning to reduce again toward the terminal end. This pattern of dynamics is in very good agreement with those seen by 13C spin relaxation. The similar signs observed for the base and sugar RDCs in the ssRNA tail and helix are consistent with the ssRNA tail adopting a conformation that is near coaxial with the helix.
To gain further insights into the dynamics of the ssRNA–helix junction, including the orientation and dynamics of the ssRNA tail relative to the reference helix, RDCs measured in the helix and ssRNA were subjected to an order tensor analysis (Losonczi et al. 1999; Bailor et al. 2007). Here, measured RDCs and an assumed local geometry for a given fragment are used to determine five elements of an order tensor describing the alignment of the fragment relative to the applied magnetic field (Saupe 1968); three Euler angles specify a principal ordering frame that describes the average orientation of the fragment relative to the applied magnetic field; a generalized degree of order (ϑ) (Tolman et al. 2001) describes the degree of fragment alignment; and an asymmetry parameter (η = Syy − Sxx/Szz) describes the asymmetry of alignment. The relative orientation of fragments can be determined by superimposing their respective order tensor frames (Losonczi et al. 1999). The relative ratio of the fragment ϑ values (ϑint = ϑSS/ϑhelix) describes the extent of interfragment motions and ranges between zero and one for maximum and minimum motional amplitudes (Tolman et al. 2001). Comparison of the fragment asymmetry values can provide insights into motional asymmetry (Tolman et al. 2001).
We determined order tensors for the helix and ssRNA fragments assuming idealized A-form helix geometry (Musselman et al. 2006). We previously showed that RDCs measured in the isolated SS tail can be well described by an A-form helical geometry (Eichhorn et al. 2012a). To further examine whether the SS tail adopts an A-form geometry when appended to a helix, we examined the agreement between the measured RDCs and values predicted using the best-fit order tensor for the following local geometries: (1) A-form helix, (2) B-form helix, (3) the average structure obtained from a previous MD simulation of the SS tail (Eichhorn et al. 2012a), and (4) X-ray structure (3FU2) and (5) NMR (2L1V) structure of the preQ1-bound RNA (Supplemental Table S3). Consistent with the 12-nt SS construct, an A-form geometry showed the best agreement with the measured RDCs. As shown in Figure 4B, for both the helical and ssRNA tail residues, we observe very good agreement between the measured RDCs and values back-predicted using the best-fit order tensor. The root mean square deviation (RMSD) between measured and predicted values is 1.5 Hz and 2.9 Hz for helix and ssRNA tail, respectively (Table 1). This compares to an RMSD, normalized to the degree of alignment of E-SS, of 0.9 Hz for the isolated SS. The order tensor elements were further examined using “leave-one-out” cross-validation. Here one measured RDC is omitted from the order tensor determination, and its value is back-predicted using the order tensor determined using all other RDCs. The process is repeated each time omitting a different RDC data point. This validation helps to identify RDCs that may strongly bias the order tensor. As shown in Figure 4B, we obtain excellent agreement in this cross-validation analysis (RMSD = 3.62 Hz) (Fig. 4B). These results suggest that both the helix and ssRNA tail assume an A-form helix-like geometry.
TABLE 1.
Summary of order tensor parameters

As expected, the helix exhibited a larger level of alignment (ϑhelix = 1.7 ± 0.07 × 10−3) compared with the more flexible ssRNA tail (ϑSS = 0.85 ± 0.1 × 10−3), yielding a small ϑint = 0.5 value between the two fragments. This small ϑint value very likely captures both collective motions of the ssRNA tail relative to the helix as well as any local motions within the ssRNA tail. As we discussed for the isolated SS construct, it is likely that any local motions in the SS tail correspond to isotropic motions due to partial melting of the stack, which result in uniform scaling of the RDCs without significantly affecting their agreement with an A-form geometry (Eichhorn et al. 2012a).
Next we determined the average relative orientation of the two fragments by superimposing their respective order tensor frames. The order tensor frame is degenerate with respect to 180° rotations about the Sxx, Syy, Szz principal directions, resulting in four distinct solutions (which we will refer to as “initial,” Sxx+ 180°, Syy+ 180°, and Szz+ 180°) for assembling two fragments (Al-Hashimi et al. 2000). We assembled the four conformations by connecting the backbone heavy atoms of residue C-1 and examined the resulting structure for steric clashes. In addition, we subjected each resulting conformation to a structure-based calculation of the order tensor using the program PALES (Zweckstetter et al. 2004; Zweckstetter 2008). Here, the overall alignment frame of the molecule is predicted based on its overall shape. The predicted alignment frames were then compared to those determined experimentally.
The Sxx+ 180° and Syy + 180° conformations are unlikely to be viable solutions because they result in steric clashes between the ssRNA tail and the helix (Fig. 5A). Moreover, they result in overall shapes and PALES-predicted order tensor frames that deviate somewhat from those determined experimentally (deviations in Szz direction are ∼4° and 7°, respectively) (Fig. 5B). Interestingly, one of the conformations (Sxx+ 180°) features the ssRNA tail possibly docked into the helix in a manner reminiscent of the conformation observed for preQ1 bound to ligand (Fig. 5A). However, such a conformation is unlikely to exist in great abundance given the very large differences in the CSs observed for residues in the ssRNA tail between the free and ligand-bound aptamer structures due to the A6 core transitioning from unpaired nucleotides to forming hydrogen bonds along the minor groove of the adjacent 3′ helix. While both the “initial” and the Szz+ 180° solutions do not lead to significant steric clashes (Fig. 5A), the “initial” conformation leads to favorable stacking between the ssRNA tail and helix, consistent with observed NOEs, and also yields slightly better agreement between measured and predicted order tensor frames (deviations in the Szz axis are 2° compared with 6°) (Fig. 5B). This conformation is characterized by average interhelical bend (βH) and twist (ζ = αH + γH) (Bailor et al. 2011) angles of ∼16° and ∼−1°, respectively, indicating that on average the ssRNA tail and the helix favor coaxial stacking with minimal interhelical twisting.
FIGURE 5.
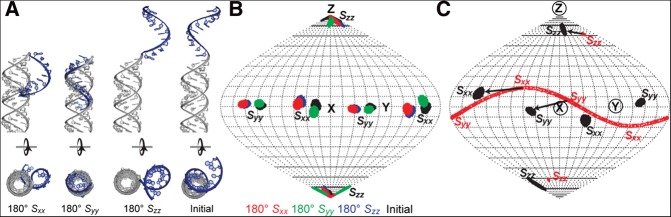
RDC-derived orientation of the helix and ssRNA tail in the ssRNA–helix junction. (A) Four degenerate orientations obtained from superimposing order tensor frames determined for the ssRNA tail and helix. (B) Sauson-Flamsteed map comparing the PALES structure-based predicted order tensor frames (Sxx, Syy, Szz) and experimental order tensor frames for each of the four degenerate solutions. (C) Comparison of the degree of alignment for the 12-nt SS (red) and 60-nt E-SS (black) constructs.
It is of interest to examine how the addition of the helix affects the overall alignment of the ssRNA tail. We previously showed that despite adopting on average an A-form-like conformation, the experimentally determined Szz axis for SS deviates from the helix axis by ∼20° (Fig. 5C; Eichhorn et al. 2012a). Such a deviation was also predicted for SS using PALES (∼14°) and was attributed to the absence of the complementary strand, which leads to an overall shape with a long axis that is not coincident with the helical axis. Interestingly, the addition of the helix to the ssRNA tail results in an Szz direction in E-SS that deviates by as little as ∼10° from the helix axis for the ssRNA (Fig. 5C). This is again consistent with the helix adopting a conformation that is nearly coaxial with the ssRNA tail, resulting in an overall shape that is more coincident with the common helix axis.
Implications for prequeuosine riboswitch function
The ssRNA sequence used in our study is located in the prequeuosine riboswitch as a 3′ overhang. On binding ligand, the ssRNA tail forms a sharp kink at the first residue (A25) at the ssRNA–helix junction site and forms a pseudoknot, where the polyadenine tract forms A-minor interactions to the helix and 3′ terminal residues base pair to the apical loop (Kang et al. 2009; Klein et al. 2009; Spitale et al. 2009; Rieder et al. 2010; Feng et al. 2011). Our studies indicate that the first two residues of the ssRNA–helix junction stack upon and extend the helix (Fig. 6). In order to form the bound conformation, A25 must unstack from the terminal helical base pair and form a sharp turn. The high level of dynamics at A27 indicates that the U26-A27 step acts as a pivot point about which the ssRNA tail moves. U-A dinucleotide steps have weak stacking energies compared with other dinucleotide steps, which may influence the dynamics at this site. A degree of flexibility near the junction site may be necessary in order to efficiently bind ligand: If the ssRNA rigidly stacks upon the helix, it may be unable to fold into the pseudoknotted structure. Conversely, if the ssRNA was very flexible in a random conformation, the large amount of accessible conformational space may inhibit efficient ligand binding. In support of the importance of the pivot point in the prequeuosine riboswitch, a pyrimidine is nearly always observed within 3 nt of the ssRNA–helix junction. A combination of a flexible junction with an ordered central region likely allows for competent binding in a timely manner while maintaining structural plasticity to rapidly adopt the bound conformation in the presence of ligand.
FIGURE 6.

Model of the proposed ligand recognition mechanism of the preQ1-I riboswitch and role of the ssRNA tail in ligand capture, revised from Eichhorn et al. (2012a).
CONCLUSIONS
ssRNA–helix junctions are ubiquitous throughout nature, yet the structural and dynamic properties of these junctions remain poorly understood. Although some studies on short 1- to 3-nt 3′ and 5′ ssRNA overhangs have reported the impact of a ssRNA overhang on helix stability, few studies have focused on the behavior of ssRNA at the end of a helix. Our data suggest that the ssRNA tail is partially ordered, on average adopting an A-form helical-like conformation that is stacked upon the helix. While the first two residues A25 and U26 stably stack upon the helix, the ssRNA tail retains a high level of dynamics, particularly at A27, indicating this residue acts as a pivot point. The degree of order increases along the central polyadenine residues, behaving similarly to the isolated 12-nt SS construct, with the degree of order approaching that of the 12-nt SS construct for the last 4 nt at the 3′ end. The appearance of additional resonances suggests conformational exchange and heterogeneity that may involve transitions between coaxial and disordered ssRNA conformations. Our results reveal a high degree of structural and dynamic complexity at the ssRNA–helix junction, which involves a fine balance between order and disorder that may facilitate efficient pseudoknot formation of the prequeuosine riboswitch on ligand recognition.
MATERIALS AND METHODS
Sample preparation
The 13C/15N A/U-labeled E-SS construct was prepared by in vitro transcription using T7 RNA polymerase as described previously (Zhang et al. 2006). The RNA was repeatedly exchanged into NMR buffer (25 mM NaCl, 15 mM sodium phosphate at pH 6.4, 0.1 mM EDTA) using an Amicon Ultra-4 (Millipore). The final RNA concentration was ∼0.4 mM. CUAC and E-SSshort constructs were purchased from Integrated DNA Technologies as a lyophilized powder at natural abundance. To prepare the sample, the RNA was dissolved in ddH2O and annealed for 5 min at 95°C followed by multiple steps of buffer exchange into NMR buffer. The final RNA concentration was ∼2 mM.
Resonance assignments
All NMR experiments were performed at 298 K, unless specified otherwise, on a Bruker Avance 600-MHz spectrometer equipped with a triple-resonance cryogenic (5-mm) probe. NMR spectra were analyzed using NMRDraw (Delaglio et al. 1995) and Sparky 3 (Goddard and Kneller 2004). The 1H, 13C, and 15N resonances in E-SS were assigned using standard homonuclear and heteronuclear 2D experiments as well as a “divide and conquer” strategy to assign helical resonances. The CUAC construct was assigned with the 1H-1H NOESY experiment using a mixing time of 250 msec. The 2D 1H-1H NOESY spectra were supplemented with 2D HCN experiments to correlate nucleobase H6/H8 to ribose C1′H1′ through the shared N1/N9 atom. Weighted CS perturbation data were calculated using the equation 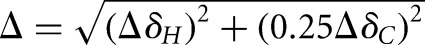 , where ΔδH and ΔδC are the CS differences in proton and carbon dimension, respectively (Cavanagh 2007).
, where ΔδH and ΔδC are the CS differences in proton and carbon dimension, respectively (Cavanagh 2007).
Carbon spin relaxation
Longitudinal (R1) and transverse (R2) carbon relaxation data for the nucleobases (C2, C6, and C8) were measured as described previously (Hansen and Al-Hashimi 2007). Due to significant differences in the relaxation properties between the elongated helix and the ssRNA, relaxation delays were optimized to each domain. The relaxation parameters used are as follows (in msec): 20, 160, 320, 400 (in duplicate), and 480 for R1 and 4, 16, 40 (in duplicate), 60 (in triplicate), and 80 (in duplicate) for R1ρ. Relaxation delays were performed in an interleaved manner with alternating short and long relaxation delays. R1 and R2 rates are listed in Supplemental Data.
The measured R1 and R2 values were used to compute relative order parameters (Lipari and Szabo 1982) using S2 = (2R2 − R1) (Dethoff et al. 2008) and normalized to yield a relative order parameter 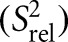 describing the relative degree of order within a molecule ranging from zero to one, where zero and one represent the minimum and maximum order, respectively. The
describing the relative degree of order within a molecule ranging from zero to one, where zero and one represent the minimum and maximum order, respectively. The 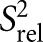 values were normalized against the helical residues with the highest values: A-30 (C8), A-40 (C2), and U-19 (C6).
values were normalized against the helical residues with the highest values: A-30 (C8), A-40 (C2), and U-19 (C6).
Measurement and order tensor analysis of RDCs
Base and sugar 1H-13C splittings were measured from the difference between the upfield and downfield components of the 1H-13C doublet along the 1H component using the narrow transverse relaxation-optimized spectroscopy (TROSY) component in the 13C dimension as implemented in 2D 1H-13C S3CT-heteronuclear single quantum correlation (HSQC) experiments (Meissner and Sorensen 1999). The measured 2H splitting was ∼8 Hz in the presence of ∼8 mg/mL Pf1 phage (Asla Biotech). Idealized A-form structures were constructed using Insight II (Molecular Simulations) correcting the propeller twist angles from +15° to −15° using an in-house program, as previously described (Bailor et al. 2007). The measured RDCs are listed in the Supplemental Data. The experimental error was estimated to be ∼3 Hz, determined by repeated measurements of the weaker helical resonances.
RDCs from the elongated helix and 3′ ssRNA tail were independently subjected to order tensor analysis using idealized A-form helices (Bailor et al. 2007). Previous NMR studies of the 12-nt ssRNA showed that the ssRNA can be modeled as an idealized helix for RDC order tensor analysis (Eichhorn et al. 2012a). Briefly, the measured RDCs and idealized A-form helices were used to determine the best-fit order tensors for both helical and ssRNA domains using singular value decomposition, implemented by the in-house written program RAMAH (Zhang et al. 2007). Another in-house program, Aform-RDC, was used to determine the order tensor errors due to inherent structural noise as well as RDC uncertainty (Musselman et al. 2006). The final RNA structure was assembled by rotating each domain into the principal axis system (PAS) of each best-fit order tensor and assembling the two helices. RDCs from tetraloop residue U-26, as well as the terminal end residues A35-A35, were excluded from analysis due to a high level of dynamics as observed in 13C spin relaxation measurements. The interhelical angles were calculated using an in-house program as previously described (Bailor et al. 2007).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
C.D.E. and H.M.A. thank members of the Al-Hashimi laboratory for insightful discussions. We thank the Michigan Economic Development Cooperation and the Michigan Technology Tri-Corridor for support in the purchase of the 600-MHz spectrometer. This work was supported by the National Institutes of Health (R21GM096156 and PO1GM0066275).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.043711.113.
REFERENCES
- Al-Hashimi HM, Valafar H, Terrell M, Zartler ER, Eidsness MK, Prestegard JH 2000. Variation of molecular alignment as a means of resolving orientational ambiguities in protein structures from dipolar couplings. J Magn Reson 143: 402–406 [DOI] [PubMed] [Google Scholar]
- Auweter SD, Oberstrass FC, Allain FH 2006. Sequence-specific binding of single-stranded RNA: Is there a code for recognition? Nucleic Acids Res 34: 4943–4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailor MH, Musselman C, Hansen AL, Gulati K, Patel DJ, Al-Hashimi HM 2007. Characterizing the relative orientation and dynamics of RNA A-form helices using NMR residual dipolar couplings. Nat Protoc 2: 1536–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailor MH, Mustoe AM, Brooks CL III, Al-Hashimi HM 2011. 3D maps of RNA interhelical junctions. Nat Protoc 6: 1536–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax A 2003. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci 12: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe JR, Nikolova EN, Eichhorn CD, Chugh J, Hansen AL, Al-Hashimi HM 2011. Characterizing RNA dynamics at atomic resolution using solution-state NMR spectroscopy. Nat Methods 8: 919–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen SJ 2009. Predicting structures and stabilities for H-type pseudoknots with interhelix loops. RNA 15: 696–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash DD, Cohen-Zontag O, Kim NK, Shefer K, Brown Y, Ulyanov NB, Tzfati Y, Feigon J 2013. Pyrimidine motif triple helix in the Kluyveromyces lactis telomerase RNA pseudoknot is essential for function in vivo. Proc Natl Acad Sci 110: 10970–10975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh J 2007. Protein NMR spectroscopy: principles and practice, p. 756 Academic Press, Boston [Google Scholar]
- Cazenave C, Uhlenbeck OC 1994. RNA template-directed RNA synthesis by T7 RNA polymerase. Proc Natl Acad Sci 91: 6972–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Starich MR, Gronenborn AM 1998. Measurement of residual dipolar couplings of macromolecules aligned in the nematic phase of a colloidal suspension of rod-shaped viruses. J Am Chem Soc 120: 10571–10572 [Google Scholar]
- Cornilescu G, Marquardt JL, Ottiger M, Bax A 1998. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc 120: 6836–6837 [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Dethoff EA, Hansen AL, Musselman C, Watt ED, Andricioaei I, Al-Hashimi HM 2008. Characterizing complex dynamics in the transactivation response element apical loop and motional correlations with the bulge by NMR, molecular dynamics, and mutagenesis. Biophys J 95: 3906–3915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey TG, Turner DH 1979. Laser temperature-jump study of stacking in adenylic acid polymers. Biochemistry 18: 5757–5762 [DOI] [PubMed] [Google Scholar]
- Eichhorn CD, Feng J, Suddala KC, Walter NG, Brooks CL III, Al-Hashimi HM 2012a. Unraveling the structural complexity in a single-stranded RNA tail: implications for efficient ligand binding in the prequeuosine riboswitch. Nucleic Acids Res 40: 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorn CD, Yang S, Al-Hashimi HM 2012b. Characterising RNA dynamics using NMR residual dipolar couplings. In Recent developments in biomolecular NMR (ed. Clore GM, Potts J), pp. 184–215 The Royal Society of Chemistry, London [Google Scholar]
- Feng J, Walter NG, Brooks CL III 2011. Cooperative and directional folding of the preQ1 riboswitch aptamer domain. J Am Chem Soc 133: 4196–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freier SM, Hill KO, Dewey TG, Marky LA, Breslauer KJ, Turner DH 1981. Solvent effects on the kinetics and thermodynamics of stacking in poly(cytidylic acid). Biochemistry 20: 1419–1426 [DOI] [PubMed] [Google Scholar]
- Fushman D, Tjandra N, Cowburn D 1999. An approach to direct determination of protein dynamics from N-15 NMR relaxation at multiple fields, independent of variable N-15 chemical shift anisotropy and chemical exchange contributions. J Am Chem Soc 121: 8577–8582 [Google Scholar]
- Getz M, Sun X, Casiano-Negroni A, Zhang Q, Al-Hashimi HM 2007a. NMR studies of RNA dynamics and structural plasticity using NMR residual dipolar couplings. Biopolymers 86: 384–402 [DOI] [PubMed] [Google Scholar]
- Getz MM, Andrews AJ, Fierke CA, Al-Hashimi HM 2007b. Structural plasticity and Mg2+ binding properties of RNase P P4 from combined analysis of NMR residual dipolar couplings and motionally decoupled spin relaxation. RNA 13: 251–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG 2004. SPARKY 3. University of California, San Francisco [Google Scholar]
- Greenfeld M, Solomatin SV, Herschlag D 2011. Removal of covalent heterogeneity reveals simple folding behavior for P4-P6 RNA. J Biol Chem 286: 19872–19879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Weiser B, Woese CR, Noller HF 1985. Comparative anatomy of 16-S-like ribosomal RNA. Prog Nucleic Acid Res Mol Biol 32: 155–216 [DOI] [PubMed] [Google Scholar]
- Hansen AL, Al-Hashimi HM 2007. Dynamics of large elongated RNA by NMR carbon relaxation. J Am Chem Soc 129: 16072–16082 [DOI] [PubMed] [Google Scholar]
- Hansen MR, Hanson P, Pardi A 2000. Pf1 filamentous phage as an alignment tool for generating local and global structural information in nucleic acids. J Biomol Struct Dyn 17 Suppl 1: 365–369 [DOI] [PubMed] [Google Scholar]
- Helm M, Brule H, Giege R, Florentz C 1999. More mistakes by T7 RNA polymerase at the 5′ ends of in vitro-transcribed RNAs. RNA 5: 618–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Peterson R, Feigon J 2009. Structural insights into riboswitch control of the biosynthesis of queuosine, a modified nucleotide found in the anticodon of tRNA. Mol Cell 33: 784–790 [DOI] [PubMed] [Google Scholar]
- Kim NK, Zhang Q, Zhou J, Theimer CA, Peterson RD, Feigon J 2008. Solution structure and dynamics of the wild-type pseudoknot of human telomerase RNA. J Mol Biol 384: 1249–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W, Hum J, Das R 2012. Ultraviolet shadowing of RNA can cause significant chemical damage in seconds. Sci Rep 2: 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DJ, Edwards TE, Ferre-D'Amare AR 2009. Cocrystal structure of a class I preQ1 riboswitch reveals a pseudoknot recognizing an essential hypermodified nucleobase. Nat Struct Mol Biol 16: 343–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeper TC, Varani G 2005. The structure of an enzyme-activating fragment of human telomerase RNA. RNA 11: 394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipari G, Szabo A 1982. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc 104: 4559–4570 [Google Scholar]
- Lodeiro MF, Filomatori CV, Gamarnik AV 2009. Structural and functional studies of the promoter element for dengue virus RNA replication. J Virol 83: 993–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczi JA, Andrec M, Fischer MW, Prestegard JH 1999. Order matrix analysis of residual dipolar couplings using singular value decomposition. J Magn Reson 138: 334–342 [DOI] [PubMed] [Google Scholar]
- Meissner A, Sorensen OW 1999. The role of coherence transfer efficiency in design of TROSY-type multidimensional NMR experiments. J Magn Reson 139: 439–442 [DOI] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman C, Pitt SW, Gulati K, Foster LL, Andricioaei I, Al-Hashimi HM 2006. Impact of static and dynamic A-form heterogeneity on the determination of RNA global structural dynamics using NMR residual dipolar couplings. J Biomol NMR 36: 235–249 [DOI] [PubMed] [Google Scholar]
- Nguyen P, Shi X, Sigurdsson ST, Herschlag D, Qin PZ 2013. A single-stranded junction modulates nanosecond motional ordering of the substrate recognition duplex of a group I ribozyme. Chembiochem 14: 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova EN, Al-Hashimi HM 2009. Preparation, resonance assignment, and preliminary dynamics characterization of residue specific 13C/15N-labeled elongated DNA for the study of sequence-directed dynamics by NMR. J Biomol NMR 45: 9–16 [DOI] [PubMed] [Google Scholar]
- O'Toole AS, Miller S, Serra MJ 2005. Stability of 3′ double nucleotide overhangs that model the 3′ ends of siRNA. RNA 11: 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole AS, Miller S, Haines N, Zink MC, Serra MJ 2006. Comprehensive thermodynamic analysis of 3′ double-nucleotide overhangs neighboring Watson–Crick terminal base pairs. Nucleic Acids Res 34: 3338–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti W, Meiler J, Bruschweiler R, Griesinger C 2002. Model-free analysis of protein backbone motion from residual dipolar couplings. J Am Chem Soc 124: 5822–5833 [DOI] [PubMed] [Google Scholar]
- Pleiss JA, Derrick ML, Uhlenbeck OC 1998. T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA 4: 1313–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollom E, Dang KK, Potter EL, Gorelick RJ, Burch CL, Weeks KM, Swanstrom R 2013. Comparison of SIV and HIV-1 genomic RNA structures reveals impact of sequence evolution on conserved and non-conserved structural motifs. PLoS Pathog 9: e1003294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder U, Kreutz C, Micura R 2010. Folding of a transcriptionally acting preQ1 riboswitch. Proc Natl Acad Sci 107: 10804–10809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saupe A 1968. Recent results in field of liquid crystals. Angew Chem Int Ed 7: 97–112 [Google Scholar]
- Schwalbe H, Buck J, Furtig B, Noeske J, Wohnert J 2007. Structures of RNA switches: insight into molecular recognition and tertiary structure. Angew Chem Int Ed Engl 46: 1212–1219 [DOI] [PubMed] [Google Scholar]
- Shi X, Solomatin SV, Herschlag D 2012. A role for a single-stranded junction in RNA binding and specificity by the Tetrahymena group I ribozyme. J Am Chem Soc 134: 1910–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Torelli AT, Krucinska J, Bandarian V, Wedekind JE 2009. The structural basis for recognition of the preQ0 metabolite by an unusually small riboswitch aptamer domain. J Biol Chem 284: 11012–11016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzfaden C, Nicholson N, Jones JJ, Guth S, Lehr R, Prescott CD, Hegg LA, Eggleston DS 2000. The structure of ribonuclease P protein from Staphylococcus aureus reveals a unique binding site for single-stranded RNA. J Mol Biol 295: 105–115 [DOI] [PubMed] [Google Scholar]
- Staple DW, Butcher SE 2005. Solution structure and thermodynamic investigation of the HIV-1 frameshift inducing element. J Mol Biol 349: 1011–1023 [DOI] [PubMed] [Google Scholar]
- Suddala KC, Rinaldi AJ, Feng J, Mustoe AM, Eichhorn CD, Liberman JA, Wedekind JE, Al-Hashimi HM, Brooks CL III, Walter NG 2013. Single transcriptional and translational preQ1 riboswitches adopt similar pre-folded ensembles that follow distinct folding pathways into the same ligand-bound structure. Nucleic Acids Res 41: 10462–10475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhang Q, Al-Hashimi HM 2007. Resolving fast and slow motions in the internal loop containing stem-loop 1 of HIV-1 that are modulated by Mg2+ binding: role in the kissing–duplex structural transition. Nucleic Acids Res 35: 1698–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra N, Bax A 1997. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278: 1111–1114 [DOI] [PubMed] [Google Scholar]
- Tolman JR, Ruan K 2006. NMR residual dipolar couplings as probes of biomolecular dynamics. Chem Rev 106: 1720–1736 [DOI] [PubMed] [Google Scholar]
- Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH 1995. Nuclear magnetic dipole interactions in field-oriented proteins: information for structure determination in solution. Proc Natl Acad Sci 92: 9279–9283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH 1997. NMR evidence for slow collective motions in cyanometmyoglobin. Nat Struct Biol 4: 292–297 [DOI] [PubMed] [Google Scholar]
- Tolman JR, Al-Hashimi HM, Kay LE, Prestegard JH 2001. Structural and dynamic analysis of residual dipolar coupling data for proteins. J Am Chem Soc 123: 1416–1424 [DOI] [PubMed] [Google Scholar]
- Tubbs JD, Condon DE, Kennedy SD, Hauser M, Bevilacqua PC, Turner DH 2013. The nuclear magnetic resonance of CCCC RNA reveals a right-handed helix, and revised parameters for AMBER force field torsions improve structural predictions from molecular dynamics. Biochemistry 52: 996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, Burch CL, Weeks KM 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460: 711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Al-Hashimi HM 2009. Domain-elongation NMR spectroscopy yields new insights into RNA dynamics and adaptive recognition. RNA 15: 1941–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Sun X, Watt ED, Al-Hashimi HM 2006. Resolving the motional modes that code for RNA adaptation. Science 311: 653–656 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Stelzer AC, Fisher CK, Al-Hashimi HM 2007. Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature 450: 1263–1267 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Kim NK, Peterson RD, Wang Z, Feigon J 2010. Structurally conserved five nucleotide bulge determines the overall topology of the core domain of human telomerase RNA. Proc Natl Acad Sci 107: 18761–18768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Kang M, Peterson RD, Feigon J 2011. Comparison of solution and crystal structures of preQ1 riboswitch reveals calcium-induced changes in conformation and dynamics. J Am Chem Soc 133: 5190–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweckstetter M 2008. NMR: prediction of molecular alignment from structure using the PALES software. Nat Protoc 3: 679–690 [DOI] [PubMed] [Google Scholar]
- Zweckstetter M, Hummer G, Bax A 2004. Prediction of charge-induced molecular alignment of biomolecules dissolved in dilute liquid-crystalline phases. Biophys J 86: 3444–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]