

In this differential RNA-seq study, Wu et al. identified 29 small RNAs expressed by the pig pathogen Streptococcus suis. The authors then went on to show that S. suis strains lacking some of these small RNAs were attenuated in a zebrafish infection model or were more sensitive to killing by pig blood. The study thus provides further evidence that small, regulatory RNAs play roles in bacterial virulence.
Keywords: small RNAs, differential RNA-seq, Streptococcus suis, TSS map, virulence
Abstract
Streptococcus suis (SS) is an important pathogen of pigs, and it is also recognized as a zoonotic agent for humans. SS infection may result in septicemia or meningitis in the host. However, little is known about genes that contribute to the virulence process and survival within host blood or cerebrospinal fluid (CSF). Small RNAs (sRNA) have emerged as key regulators of virulence in several bacteria, but they have not been investigated in SS. Here, using a differential RNA-sequencing approach and RNAs from SS strain P1/7 grown in rich medium, pig blood, or CSF, we present the SS genome-wide map of 793 transcriptional start sites and 370 operons. In addition to identifying 29 sRNAs, we show that five sRNA deletion mutants attenuate SS virulence in a zebrafish infection model. Homology searches revealed that 10 sRNAs were predicted to be present in other pathogenic Streptococcus species. Compared with wild-type strain P1/7, sRNAs rss03, rss05, and rss06 deletion mutants were significantly more sensitive to killing by pig blood. It is possible that rss06 contributes to SS virulence by indirectly activating expression of SSU0308, a virulence gene encoding a zinc-binding lipoprotein. In blood, genes involved in the synthesis of capsular polysaccharide (CPS) and subversion of host defenses were up-regulated. In contrast, in CSF, genes for CPS synthesis were down-regulated. Our study is the first analysis of SS sRNAs involved in virulence and has both improved our understanding of SS pathogenesis and increased the number of sRNAs known to play definitive roles in bacterial virulence.
INTRODUCTION
Streptococcus suis (SS) is considered to be a major pathogen worldwide in the swine industry and is also an important zoonotic agent for people in contact with infected pigs or pork-derived products (Gottschalk and Segura 2000). Among the 33 serotypes described, SS serotype 2 (SS2) is most frequently associated with a range of diseases including meningitis, pneumonia, septicemia, and arthritis (Hill et al. 2005; Gottschalk et al. 2010). During an outbreak of SS in China in 2005, 39 out of the 215 patients infected with SS died as a result of the infection (Yu et al. 2006). Furthermore, SS has been reported to be the first and second most common cause of adult meningitis in Vietnam and Thailand, respectively (Fittipaldi et al. 2009). It colonizes epithelial cells of the upper respiratory tract and accesses the blood or directly enters the bloodstream through cuts in the skin (Gottschalk and Segura 2000; Gottschalk et al. 2010). Once in the bloodstream, bacteria traveling in the blood may result in bacteremia or septicemia. When reaching the blood-brain barrier (BBB), bacteria can cross it and enter the cerebrospinal fluid (CSF), which may result in meningitis (Gottschalk and Segura 2000; Gottschalk et al. 2010). Bacteria can multiply freely within the CSF and spread over the entire surface of the brain (Small et al. 1986; Scheld et al. 2004). Bacterial components are released by replication and autolysis of bacteria in the CSF, which triggers the inflammatory response in the subarachnoid space by inducing the production of inflammatory cytokines and chemokines (Scheld et al. 2004). Clinical outcome in bacterial meningitis is directly associated with the number of bacteria and the concentrations of bacterial components in the CSF (Schneider et al. 1999; van der Flier et al. 2003).
To cause infections, SS regulates the expression of virulence factors in an exquisite manner. Research on gene regulation has focused mainly on protein regulators such as two-component or stand-alone regulators, including LuxS (Cao et al. 2011), CcpA (Willenborg et al. 2011), CovR (Pan et al. 2009), CiaR/H (Li et al. 2011), Ihk/Irr (Han et al. 2012), AdcR (Aranda et al. 2009b), ArgR (Fulde et al. 2011), SalK/R (Li et al. 2008), RevSC21 (Wu et al. 2009), and Rgg (Zheng et al. 2011). Recent studies, however, have revealed that regulatory RNAs (small RNAs from intergenic regions [sRNAs] and antisense RNA [asRNA]) have emerged as key regulators of virulence in several bacteria (Romby et al. 2006; Toledo-Arana et al. 2009; Waters and Storz 2009; Sharma et al. 2010; Wurtzel et al. 2012). However, none have been confirmed by experimental approaches in SS. To understand how SS responds within pig blood and pig CSF, we investigated its transcriptome using differential RNA-sequencing (dRNA-seq). This approach, selective for the 5′ ends of primary transcripts, has recently been used for transcriptome analysis, transcriptional start site (TSS) determination, and regulatory RNA discovery in many organisms (Albrecht et al. 2010; Mitschke et al. 2010; Sharma et al. 2010). In the present study, we depicted the transcriptional landscape of SS within pig blood and CSF, revealed, for the first time, the SS sRNAs involved in virulence, and increased the pool of sRNAs that play definitive roles in bacterial virulence.
RESULTS
Characteristics of sequencing data
We compared the transcripts from SS strain P1/7, a virulent strain isolated from a pig dying from meningitis (Holden et al. 2009), grown to exponential phase at 37°C in rich medium (as a control condition [PC]) with two ex vivo conditions: incubating bacteria in pig blood (PB) and pig CSF (PCSF) for 60 min. For each condition, two differential cDNA libraries were sequenced: One library, denoted (+), was enriched for primary transcripts by Terminator 5′-Phosphate-Dependent Exonuclease (TEX) that degrades 5′ P such as mature rRNA, tRNA, and processed transcripts, but not 5′ PPP RNA, to include most mRNAs and regulatory RNAs (TSS sequencing [TSS-seq]); and the other cDNA library, denoted (−), from bacterial RNA without TEX treatment (strand-specific sequencing [RNA-seq]). In total, we obtained ∼1.83 Gbp data and 42 M 50-bp reads for RNA-seq and TSS-seq, respectively. For TSS-seq, the 23S and 16S rRNAs were significantly reduced after TEX treatment (Fig. 1A). The enriched RNAs were the transcripts with complete TSS information and were sequenced by high-throughput sequencing. The sequenced reads were uniquely mapped to the genome and compared with the RNA-seq data. For example, the cDNA reads with TEX treatment were clustered at the 5′ end of SSU0939, whereas the cDNA reads without TEX treatment were uniformly distributed over SSU0939 (Fig. 1B).
FIGURE 1.

Characteristics of sequencings. (A) Analysis of RNA before (left) and after (right) TEX treatment using a Bioanalyzer 2100. The peaks of 23S and 16S are significantly declined after TEX treatment, so the fluorescence absorption signals of the RNAs become weak. (B) The cDNA reads with (+) or without (−) TEX treatment mapped to the SS genome. The coding sequence and 5′ UTR of SSU0939 are indicated by blue and black bars, respectively, and the arrows on the blue bar show the direction of the gene.
Genome-wide mapping of SS TSSs
A total of 793 TSSs were defined in this organism (Supplemental Table S1), and the median length of the 5′ untranslated region (UTR) is 28 nt (Fig. 2A). Further analysis of sequences upstream of the determined TSSs revealed 91% (589/647) primary and secondary TSSs with extended Pribnow boxes (tgnTAtaAT) as −10 motifs and a periodic AT-rich signal as −35 motifs (Fig. 2B). Seventy-seven leaderless mRNAs (5′ UTR < 10 nt in Supplemental Table S1) were found. Many of the corresponding genes were presumed to have housekeeping functions, as has been described in Helicobacter pylori (Sharma et al. 2010) and the plant pathogen Xanthomonas (Schmidtke et al. 2012). In addition to riboswitches, some surface proteins and virulence-associated factors, including muramidase-released protein (MRP) and extracellular protein factor (EF), have long 5′ UTRs (5′ UTR > 70 nt in Supplemental Table S1), similar to those found in Listeria monocytogenes (Loh et al. 2006; Wurtzel et al. 2012). In L. monocytogenes, the post-transcriptional control by the long 5′ UTR is important for bacteria virulence (Loh et al. 2006).
FIGURE 2.

TSS annotation and 5′ UTRs. (A) 5′ UTR lengths. Frequency of individual 5′ UTR lengths based on 647 TSSs (primary and secondary) of mRNAs. (B) Promoter motif. Motif searches upstream of SS TSSs revealed extended Pribnow boxes (−10 signal: tgnTAtaAT), of which 589/647 (∼91%) were primary and secondary (top, histogram of distribution; bottom, logo of upstream TSS sequences). The dotted line indicates the upstream periodic AT-stretch signal. (C) Re-annotation of the SSU0647 gene prompted by a newly discovered TSS downstream from its originally annotated start codon. (Upper) The alignment of protein sequences of Streptococcus strains (Supplemental Table S2). (Lower) Reads of PC+/PC− libraries mapping to the operon SSU0647-SSU0645 are shown. The highly conserved re-annotated new AUG and −10 box of the new TSS are shown.
The unambiguous mapping of TSSs and direct visualization of transcribed genomic loci enable optimization of the SS genome annotation. Based on a primary TSS, located within the previously annotated open reading frame (ORF), and the conservation of the start codon in other Streptococcus species (Supplemental Table S2) downstream from the primary TSS, we proposed corrections for four genes (Supplemental Table S3). We took SSU0647 as an example. As shown in Figure 2C, the SSU0647 primary TSS lying within the previously annotated start codon (old start codon) and the sequences conserved among Streptococcus species suggested that the new start codon could be located 6 bp downstream from the old start codon. To validate the new start codon, a combination of a green fluorescent protein (GFP) tag and frame-shift mutagenesis was used, according to a previous report (Smollett et al. 2009). The SSU0647 nucleotide sequence, from 200 bp upstream of the annotated start codon to 33 bp downstream from the new start codon, was fused to GFP (starting from the second residue of GFP) (Supplemental Fig. S1). The fused fragment was cloned into pSET4s, and then the fusion plasmid (0647::GFP) was transformed into SS P1/7. Two single-nucleotide deletions were created: 0647::GFP-m1 (the “A” base just downstream from the old ATG was deleted) and 0647::GFP-m2 (the “A” base just downstream from the new ATG was deleted; marked in red and numbered in Supplemental Fig. S1). Western blot analysis of SS P1/7 harboring pSET4s, 0647::GFP, 0647::GFP-m1, and 0647::GFP-m2 fusion plasmids was performed using anti-GFP. A band corresponding to 0647::GFP was detected in SS P1/7 harboring 0647::GFP and 0647::GFP-m1 fusion plasmids but was not detected for the pSET4s or 0647::GFP-m2 fusion plasmid (Supplemental Fig. S1). Therefore, SSU0647 is not translated from the old start site and is, instead, translated from a new start codon.
SS operon map
In prokaryotic organisms, functionally related genes are organized into operons. Knowledge of the operons is important for deciphering gene function. To present the operon map for SS, we identified 370 operons encoding 1195 genes that represent 60.7% of all SS annotated genes by combining TSS-seq data with RNA-seq analyses (Supplemental Table S4; Supplemental Fig. S2A–C). Comparing with the database of prokaryotic operons (DOOR) (Mao et al. 2009), 276 operons predicted by the DOOR have been confirmed. The operons that were differentially expressed in blood or CSF are summarized in Supplemental Table S4.
The genes in operon 108 (from SSU0515 to SSU0523) that are involved in the biosynthesis of capsular polysaccharide (CPS) (Holden et al. 2009) were up-regulated in blood, compared to the control condition (Fig. 3A). The ability of SS to resist phagocytosis and persist in blood largely depends on the production of CPS. However, these genes were down-regulated in CSF. Dot-blot analysis was performed using anti-serotype 2 polyclonal antibody absorbed by a P1/7 CPS-deficient mutant, which was generated by transposon mutagenesis (C Ju and C Lu, unpubl.), to further quantify the amount of CPS produced by SS under the different experimental conditions, according to previous reports (Rubens et al. 1987; Chaffin et al. 2000, 2005). Dot-blot signals were increased in blood and decreased in CSF, compared to the control condition (Fig. 3B). These data suggested that the amount of CPS was increased in blood and decreased in CSF, which is in agreement with the results obtained by dRNA-seq. The genes in operon 109 (from SSU0535 to SSU0540) for the biosynthesis of sialic acid, a major element of bacterial surface structure, were up-regulated in blood. Operons 108 and 109 were validated by RT-PCR (Supplemental Fig. S2D).
FIGURE 3.

CPS operon. (A) The genes in operon 108, from SSU0515 to SSU0523, for the biosynthesis of CPS. Reads from the control condition (PC+/−), pig blood (PB+/−), and pig CSF (PCSF+/−) libraries mapped onto CPS operon annotations in positive and negative strands (genome position at the bottom). Scales (0–200) give normalized depths (the number of mapped clean reads for each gene was counted and then normalized to the RPKM value). (B) Dot-blot evaluating the amount of CPS using anti-serotype 2 polyclonal antibody absorbed by P1/7 CPS-deficient mutant. Dilutions of cells were made in PBS and spotted onto nitrocellulose membranes (1 = 106 CFU); the cells were then fixed and the membranes were probed with the absorbed antibody. A P1/7 CPS-deficient mutant generated by transposon mutagenesis was used as a control.
SS regulatory RNA
TSSs at intergenic regions were analyzed to detect the trans-encoded sRNAs in SS. In total, 29 intergenic transcripts were identified in the SS genome (Fig. 4A; Supplemental Table S5). dRNA-seq can accurately identify TSSs on both strands of the DNA. The strand specificity of the TSS mapping allowed us to identify asRNA in SS. In total, 40 asRNAs were detected in SS (Supplemental Table S6). Two sRNAs (rss04 and rss05) were verified by modified rapid amplification of cDNA ends (RACE), allowing simultaneous determination of 5′ and 3′ ends (Toledo-Arana et al. 2009). The RACE results correlated with the dRNA-seq data (Fig. 4B,C). Eight sRNAs were verified by Northern blot analysis (Fig. 4B; Supplemental Fig. S3). The known sRNAs predicted by the Rfam database (Burge et al. 2012) against the SS P1/7 genome, such as 6S RNA, tRNAs, rRNAs, tmRNA, and RNase P, were confirmed to be expressed in this study. Four sRNAs identified in this study, rss10, rss21, rss16, and rss29 were annotated in the Rfam database as Lacto-rpoB RNA, THF riboswitch, RNase P, and tmRNA, respectively. Four regulatory RNAs are predicted to contain small ORFs (ORF sizes from 22 to 46 aa) (Supplemental Table S7). Homology searches revealed that 10 sRNAs (rss08, rss10, rss13, rss16, rss19, rss21, rss24, rss25, rss27, and rss29) are also found in other pathogenic Streptococcus species, including S. equi subsp. Zooepidemicus, S. pneumoniae, S. pyogenes, and S. agalactiae (Fig. 5). The 29 sRNA sequences are presented in Supplemental Table S5, and the secondary structure of 15 sRNAs (rss03, rss04, rss05, rss06, rss11, and 10 conserved sRNAs) predicted by RNAfold are shown in Supplemental Figure S4.
FIGURE 4.

SS sRNA. (A) Genomic map showing the localization of SS sRNA. (B) rss04 in different conditions, showing it is located between SSU0908 and SSU0909. (C) rss05 in different conditions, showing it is located between SSU1064 and SSU1065. (Left) Annotated ORFs are indicated by blue bars, with arrows showing the direction of the gene. (Right) RACE results are shown, and they correlate with dRNA-seq data. Scales give normalized depths (the number of mapped clean reads for each gene was counted and then normalized to the RPKM value). Northern blot analysis of rss04 was performed using the RNA extracted from SS in the control condition.
FIGURE 5.

The presence of homologs of SS sRNAs in other streptococcal strains. Homology searches revealed that 10 sRNAs (rss08, rss10, rss13, rss16, rss19, rss21, rss24, rss25, rss27, and rss29) were also found in other pathogenic Streptococcus species. Red indicates the sRNAs are present, and green indicates the sRNAs are absent in other streptococcal strains.
Deletion of sRNAs attenuating SS virulence in a zebrafish infection model
sRNAs were next analyzed for their potential role in virulence. We hypothesized that genes with enhanced expression in response to incubation in pig blood or CSF might contribute to SS virulence. Three sRNAs (rss05, rss06, and rss12) were up-regulated in blood, and six sRNAs (rss03, rss04, rss06, rss08, rss11, and rss16) were up-regulated in CSF, raising the possibility that they might be involved in virulence. Six sRNA deletion mutants (Δrss03, Δrss04, Δrss05, Δrss06, Δrss11, and Δrss12) were generated successfully. RT-PCR results confirmed the presence of sRNAs in the wild-type strain P1/7 and complemented strains and the absence of sRNAs in deletion mutants (Supplemental Fig. S5A). The sRNAs were deleted with no polar effects on flanking genes (Supplemental Fig. S5B). The virulence of the six sRNA mutants and P1/7 wild-type strain were evaluated in a zebrafish model. Zebrafish has been shown to be a useful model for evaluating the virulence of SS (Wu et al. 2010b; Wang et al. 2011; Ju et al. 2012; Tang et al. 2013). The sRNA deletion mutants Δrss03, Δrss04, Δrss05, Δrss06, and Δrss11 were significantly less virulent than the P1/7 wild-type strain, and the virulence of mutants was restored in complemented strains (Fig. 6).
FIGURE 6.

Evaluating the virulence of sRNA deletion mutants in a zebrafish model. Zebrafish were challenged with wild-type strain P1/7, sRNA deletion mutants, and sRNA complemented strains. The LD50 was calculated to compare the virulence of strains. An unpaired t-test was used to compare log10LD50 between strains P1/7 and Δrss12. For the remaining comparisons, statistical analyses were performed using one-way ANOVA and Dunnett's multiple comparisons test. (*) P< 0.05, (**) P< 0.01. C-rss03, C-rss04, C-rss05, C-rss06, and C-rss11 denote the complemented strains for rss03, rss04, rss05, rss06, and rss11, respectively.
SS sRNAs contribute to bacterial survival in pig blood
We next incubated the P1/7 wild-type and mutant strains in pig whole blood for 4 h, and samples were taken to assess survival through CFU determination. Compared with the wild-type strain P1/7, rss03, rss05, and rss06 deletion mutants were significantly more sensitive to killing by pig blood (Fig. 7A). The sRNA mutants and wild-type strain P1/7 were also characterized for their growth in Todd-Hewitt broth (THB) at 37°C under the same experimental conditions used for the incubation in blood. The growth rate for the mutants was comparable to that of the P1/7 wild-type strain (Fig. 7B), indicating that the phenotype observed in blood was not due to a growth defect.
FIGURE 7.
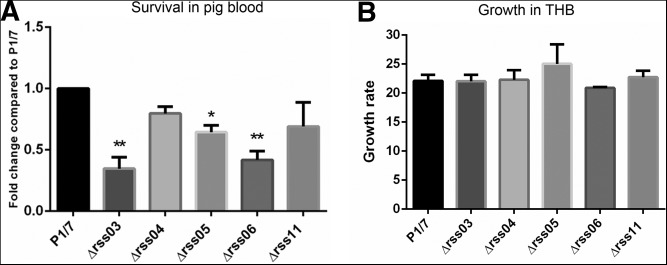
Survival of sRNA deletion mutants in pig blood. (A) Survival of wild-type stain P1/7 and sRNA deletion mutants in pig blood. Wild-type stain P1/7 and sRNA deletion mutants were tested for survival using the ex vivo pig blood model over a time course of 4 h. The survival results were expressed as a fold change relative to the level of wild-type strain P1/7. (B) sRNA mutants and wild-type strain P1/7 were also characterized for their growth in THB at 37°C under the same experimental conditions. Growth rate was calculated as CFU at 4 h/CFU at 0 h. Statistical analyses were performed using one-way ANOVA and Dunnett's multiple comparisons test. (*) P< 0.05, (**) P< 0.01.
rss03 and rss06 are involved in regulating virulence factor genes
We next investigated how these sRNAs regulate SS virulence. rss03 was predicted to interact with SSU0516 (cps2B), which is involved in the biosynthesis of CPS (Supplemental Fig. S6A). Compared with wild-type strain P1/7, levels of cps2B and cps2C (SSU0517) mRNA expression were increased 2.8-fold in Δrss03, and their expression was restored in a rss03 complemented strain, confirming that rss03 repressed their expressions (Supplemental Fig. S6A). In Δrss06, a virulence gene SSU0308 encoding a zinc-binding lipoprotein was decreased 6.4-fold compared with the wild-type strain, and it was fully restored by complementation, indicating that rss06 positively affects SSU0308 expression (Supplemental Fig. S6B). ΔG values, calculated by the RNAhybrid program (Rehmsmeier et al. 2004; Loh et al. 2009), for rss03-SSU0516 and rss06-SSU0308 pairings were −23.5 kcal/mol and −19.9 kcal/mol, respectively. To test whether rss03 directly regulates SSU0516 and rss06 directly regulates SSU0308, several base substitutions that increased the theoretical ΔG value were introduced into the predicted pairing region of rss03 (rss03-m3, theoretical ΔG increasing from −23.5 to −12.4 kcal/mol) and the predicted pairing region of rss06 (rss06-m6, theoretical ΔG increasing from −19.9 to −11.3 kcal/mol) (Supplemental Fig. S6A,B). Next, rss03-m3 and rss06-m6 were complemented into Δrss03 (C-rss03-m3) and Δrss06 (C-rss06-m6), respectively. If they directly regulate their targets by these putative pairings, such base substitutions should weaken the complementation effect. However, the complementation effect for C-rss03-m3 and C-rss06-m6 was similar to that for C-rss03 and C-rss06, respectively (Supplemental Fig. S6A,B). Moreover, we assessed the putative base-pairing interaction using a GFP-based pXG plasmid system (Urban and Vogel 2007). This system has been used successfully to verify sRNA-mRNA interactions in many bacteria, including Escherichia coli (Urban and Vogel 2007), Salmonella enterica serovar Typhimurium (Sharma et al. 2007), S. agalactiae (Pichon et al. 2012), and Prochlorococcus marinus (Richter et al. 2010). Since SSU0516 is inside an operon, a 0516::GFP fusion was constructed with the pXG-30 plasmid which is particularly suitable for a gene within an operon. SSU0308 is a monocistronic gene, so a 0308::GFP fusion plasmid was constructed with the pXG-10 plasmid, which is suitable for a monocistronic gene (Urban and Vogel 2007). Colony fluorescence experiments showed that E. coli strains harboring the 0516::GFP plasmid with either the control plasmid or rss03 expression plasmid showed similar fluorescence. A similar fluorescence was also observed in E. coli strains harboring the 0308::GFP fusion plasmid with either the control plasmid or rss06 expression plasmid (Supplemental Fig. S6C). We also performed Western blot analysis using anti-GFP, which showed similar signals in E. coli strains harboring the 0516::GFP fusion plasmid with either the control vector or rss03 expression plasmid, and 0308::GFP fusion plasmid with either the control vector or rss06 expression plasmid (Supplemental Fig. S6D). These data suggested that rss03 indirectly regulates SSU0516 and rss06 indirectly regulates SSU0308.
Since SSU0516 and SSU0517 were increased 2.8-fold in Δrss03, Dot-blot analysis was performed to evaluate whether the amount of CPS increases in Δrss03. rss03 is expressed at a high level in three conditions (Supplemental Fig. S3A). Thus, the capsule assays for the wild-type strain P1/7 and ▵rss03 were carried out in the control condition. The Dot-blot revealed similar signals between Δrss03 and the wild-type strain P1/7 (Supplemental Fig. S7A), indicating that they each have a similar amount of CPS. rss03 contributes to SS survival in blood, so it may be involved in regulating genes that are important for SS survival in blood. We found that one of these genes, sspA, was decreased fivefold in Δrss03 and the expression of sspA was restored in the rss03 complemented strain (Supplemental Fig. S7B). sspA encodes a surface-associated subtilisin-like protease, which is capable of degrading IL-8, a major chemo-attractant of neutrophils (Fittipaldi et al. 2012). Deletion of sspA attenuated SS virulence in a mouse model and was more susceptible to killing by whole blood than the wild-type strain P1/7 (Bonifait et al. 2010). This could be a potential way through which rss03 contributes to the survival of SS in blood.
Transcriptional changes in infection conditions
Compared with the control condition, in blood, there were 898 genes differentially expressed with a difference greater than twofold (Supplemental Table S8). In total, 400 genes were up-regulated, including genes involved in subversion of the host defenses. In CSF, SS encounters a variety of nutritional limitations and stresses. Comparison of gene expression levels between CSF and control conditions revealed an extensive transcriptional reshaping, with 268 up-regulated genes and 728 down-regulated genes (Supplemental Table S9). In accordance with gene expression studies on other meningitis-associated bacteria in CSF, many of the up-regulated genes were related to the transport and metabolism of carbohydrates, amino acids, and nucleotides (Fig. 8).
FIGURE 8.
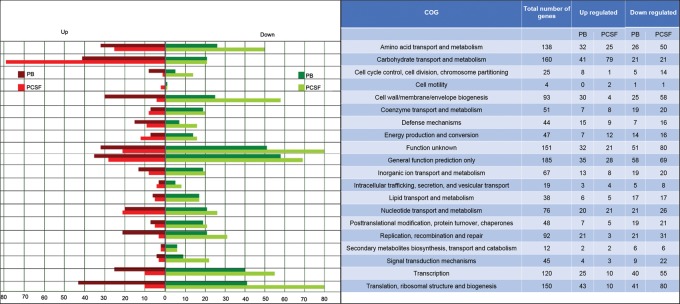
Gene expression analysis. (Left) Bars indicate the numbers of genes with modified expression in PB or PCSF relative to PC. (Right) Genes were classified into 20 main functional categories in the COG database.
To confirm the expression of genes obtained by RNA-seq experiments, quantitative real-time PCR (qRT-PCR) was performed on 13 genes (Fig. 9A) and nine regulatory RNAs—seven sRNAs and two asRNAs (Fig. 9B). The qRT-PCR results produced similar log2 ratios to the corresponding RNA-seq results. Thus, the qRT-PCR analysis confirmed the reliability of the transcriptional data obtained from the RNA-seq experiments.
FIGURE 9.
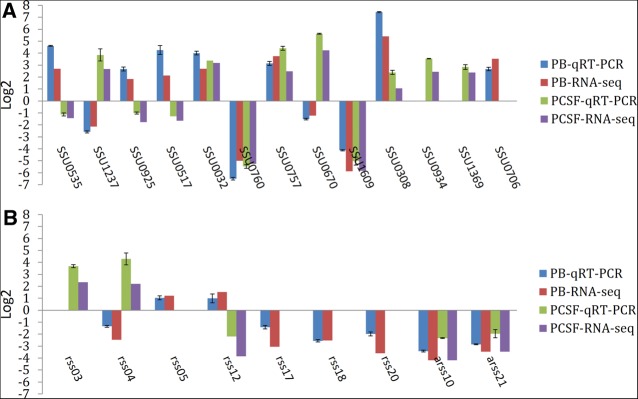
Validation of gene and regulatory RNAs expression by qRT-PCR. To verify the data obtained by RNA-seq, qRT-PCR was performed on 13 genes and nine regulatory RNAs. (A) Gene expression in blood and CSF. (B) Regulatory RNAs expression in blood and CSF.
DISCUSSION
The dRNA-seq analysis of the SS transcriptome under different experimental conditions led to remarkable insights into the transcriptional landscape of this important swine pathogen. In addition to the SS TSS map, we have provided an annotation of 5′ UTRs and a re-annotation of reading frames for four genes. Our results represented the first SS operon map in various environments, which has helped us to understand the adaptation mechanisms of SS when the bacterium enters the host.
Numerous sRNAs have been identified in bacterial pathogens, which carry out a variety of biological functions, including metabolism, stress adaptation, and virulence. Many sRNAs have been recently identified in streptococcal species, including S. pyogenes (Deltcheva et al. 2011; Tesorero et al. 2013), S. pneumoniae (Kumar et al. 2010; Mann et al. 2012), S. mutans (Lee and Hong 2012), and S. agalactiae (Pichon et al. 2012). Several of these sRNAs in streptococcal species have been reported to regulate virulence. In S. pyogenes, six sRNAs (FasX, Pel, RivX, 4.5S RNA, tracrRNA, and crRNAs) were demonstrated to regulate virulence (Le Rhun and Charpentier 2012; Tesorero et al. 2013). Mann et al. (2012) identified several sRNAs with global and niche-specific roles in virulence in S. pneumoniae. In the present study, we identified 29 SS sRNAs by dRNA-seq, five of which were involved in virulence. Ten sRNAs were predicted to exist in many streptococcal bacteria (Fig. 5). Since rss10 (Lacto-rpoB RNA), rss16 (RNase P), and rss29 (tmRNA) were annotated as known RNA families described in the Rfam database, it was not surprising that these sRNAs were predicted to be present in most of the streptococcal bacteria investigated. Tesorero et al. (2013) reported that six sRNAs from the S. pyogenes strain MGAS315 were predicted to be present in other streptococci. Two of them (SSRC10 and SSRC29) were predicted to exist in SS strain BM407, but they were not detected in the present study. S. pyogenes sRNA SSRC32, annotated as RNase P, was detected in the present study (rss16). Secondary structure of 15 sRNAs predicted by RNAfold is shown in Supplemental Figure S4, which might help to identify particular structure or sequence motifs that are involved in sRNA-mRNA interactions. For instance, rss24 was predicted to carry a stem–loop structure with a conserved C-rich sequence in the apical loop (Supplemental Fig. S4L). The stem–loop structure contains C-rich sequences in the loops that are likely involved in base-pairing with G-rich ribosome binding sites (RBS) (Vanderpool et al. 2011). Staphylococcus aureus sRNA RNAIII uses C-rich sequences in their apical loops base-pairing with RBS of target coa, spa, SA1000, and SA2353 mRNAs, resulting in preventing ribosome binding and translation initiation (Huntzinger et al. 2005; Boisset et al. 2007; Chevalier et al. 2010; Vanderpool et al. 2011). S. aureus sRNA SprX inhibits SpoVG expression through the direct interaction between a C-rich loop of SprX and RBS of target mRNA (Eyraud et al. 2014). The conservation of the C-rich loop of rss24 within the Streptococcus species suggests a common function. Additionally, for these 10 conserved sRNAs, the regions that are conserved within the Streptococcus species have been shown by different colors (Supplemental Fig. S4E,F, H–O). These Streptococcus pathogens can infect different hosts; it is, therefore, plausible to hypothesize that these sRNAs might be involved in the global adaptation processes during infection.
In the present study, we showed that rss03 indirectly repressed the expression of cps2B and cps2C. In CSF, cps2B and cps2C were down-regulated, whereas rss03 was up-regulated, when compared with the wild-type strain. This has led us to hypothesize that rss03 could help to down-regulate the expression of cps2B and cps2C when SS reaches the CSF during an infection. We indicated that rss06 indirectly activates SSU0308 expression. SSU0308, which encodes a zinc-binding lipoprotein, has been reported as a virulence factor (Aranda et al. 2012). The virulence of the SSU0308 deletion mutant was more than 50 times lower than that of the wild-type strain P1/7 in a mouse model (Aranda et al. 2012). This lipoprotein might be a promising candidate for vaccines against SS infection (Aranda et al. 2008, 2012). Both SSU0308 and rss06 were up-regulated in blood and CSF. These results suggested that one of the mechanisms through which rss06 contributes to SS virulence is by activating SSU0308 expression indirectly. Because rss03 and rss06 are not in the list of 10 conserved sRNAs (Fig. 5), rss03 and rss06 may be only found in S. suis.
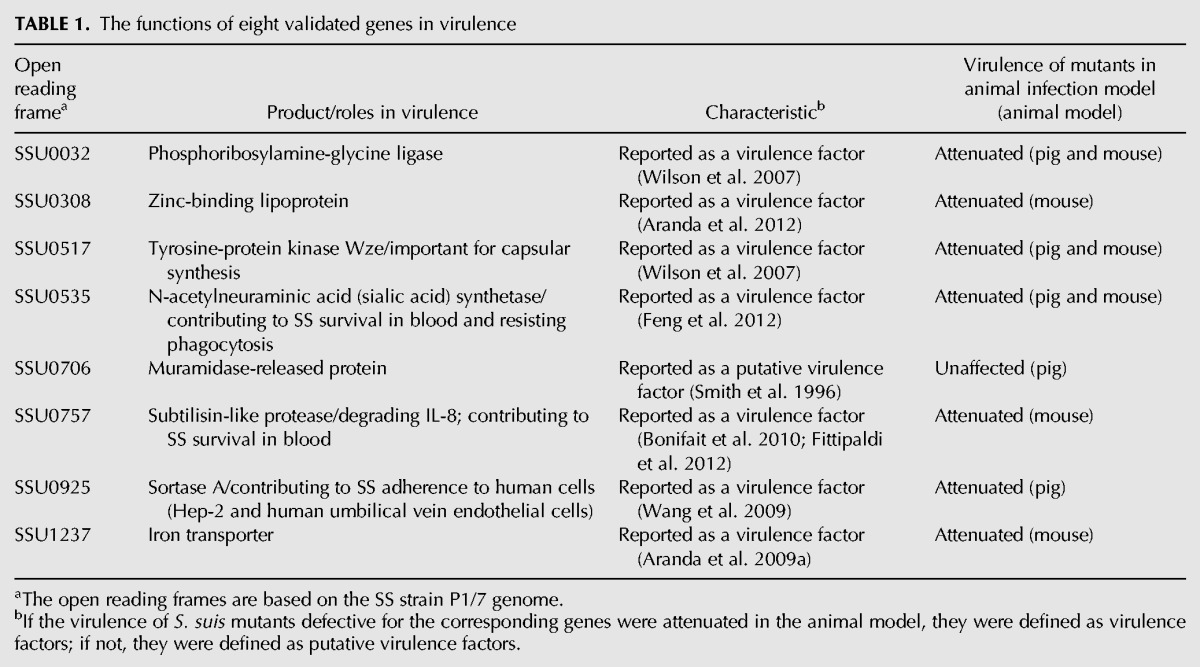
Despite its importance in disease, relatively little is known about how SS adapts to survive in pig blood. In addition, SS encounters a range of other nutritional limitations and stresses in host fluids, such as CSF, during systemic infection. The differential expression of genes in blood or CSF not only contributes to the understanding of how SS survives and proliferates within the host environment, but it could also facilitate the development of vaccines or therapeutic agents against SS infection. Eight of the 13 genes that were validated by qRT-PCR were reported as virulence-associated factors. The functions of these genes in virulence are summarized in Table 1. In blood, seven of the eight genes were up-regulated, including the genes in operon 109 for the biosynthesis of sialic acid. Overexpression of sialic acid in the blood has been shown to be required for avoiding phagocytosis and killing by innate immune cells (Fittipaldi et al. 2012). The up-regulation of genes for the biosynthesis of sialic acid, together with the phenotype of decreased survival that was observed for the deletion mutant in the ex vivo whole blood model (Feng et al. 2012), confirmed the important role that sialic acid plays in resistance to whole-blood killing in the host environment. Two genes encoding immunoglobulin proteases were also up-regulated in blood: iga (SSU0879) that encodes IgA1 protease, deletion of which has been shown to attenuate SS virulence in a pig model (Zhang et al. 2011); and ideSsuis (SSU0496) that encodes IgM-specific protease, of which the isogenic mutant ΔideSsuis was significantly attenuated in survival in pig blood compared with the wild-type strain (Seele et al. 2013). In CSF, we showed that genes in operon 108 involved in CPS synthesis were down-regulated compared with the control condition. In agreement with our study, Tuomanen et al. (1985) reported that the presence of CPS in S. pneumoniae was not necessary for bacterial growth in CSF. Many studies on CPS-deficient mutants have clearly shown bacterial cell wall components to be the major cytokine-inducing factors (Fittipaldi et al. 2012). Lecours et al. (2011) showed that CPS impairs cytokine release by hindering cell wall components; thus, reduced levels of CPS could result in exposing bacterial cell wall components and facilitate an increased inflammatory response. Studies on meningitis-associated bacteria have shown that a fatal disease outcome can be caused by the host inflammatory response (Dominguez-Punaro et al. 2007). Dominguez-Punaro et al. (2010) showed that after being stimulated by an SS CPS-deficient mutant, the microglia induced significantly higher levels of proinflammatory mediators than that of the wild-type strain. Here, our results suggest that SS increases CPS as a protective mechanism against phagocytosis and killing in blood and reduces expression of CPS in CSF to expose the cell wall components, thereby facilitating a higher inflammatory response in the brain. When SS encountered poor nutritional conditions in CSF, many genes involved in the transport and metabolism of carbohydrates, amino acids, and nucleotides were up-regulated, presumably to promote SS proliferation. SSU0318, which encodes a universal stress protein, was up-regulated in CSF, which may indicate that the bacteria are under stress in adapting from the rich medium to the poor medium of the CSF.
TABLE 1.
The functions of eight validated genes in virulence
In this paper, we have presented a genome-wide map of TSSs and operons in various environments, paving the way to the understanding of how SS adapts to survive and grow in the host blood or CSF. In addition, we showed that five sRNAs contribute to SS virulence. Our study is the first to identify SS sRNAs, provides the first evidence for a role of sRNAs in SS pathogenesis, and has increased the number of sRNAs that play definitive roles in bacterial virulence.
MATERIALS AND METHODS
Bacterial strains and growth conditions
Bacterial strains and plasmids used in this study are listed in Supplemental Table S10. SS virulent strain P1/7 was originally isolated from a pig dying of meningitis (Holden et al. 2009). All SS strains were grown in Todd-Hewitt broth (THB; Oxoid) and plated on Todd-Hewitt agar (THA) at 37°C. E. coli strains were cultured in Luria-Bertani (LB; Becton Dickinson) medium at 37°C. When required, the following antibiotics were added to the medium at the indicated concentrations: for SS, spectinomycin at 100 μg/mL; for E. coli, spectinomycin at 50 μg/mL, ampicillin at 100 μg/mL, or chloramphenicol at 25 μg/mL.
Bacteria incubation in pig blood or CSF
Blood and CSF samples were collected from healthy pigs that tested negative for SS serotype 2, as determined by ELISA (Zhang and Lu 2007). SS strain P1/7 was cultured to the exponential growth phase (OD600 nm ∼0.6). Bacteria were centrifuged at 8000 rpm for 5 min, washed in PBS, then resuspended in an equal volume of freshly isolated pig blood or CSF, and incubated for 60 min at 37°C with occasional gentle shaking. Two volumes of RNAprotect Bacteria Reagent (Qiagen) were added to each sample according to the manufacturer's instructions. For collecting bacteria from blood, the tubes were centrifuged at 1000 rpm for 5 min to remove blood cells, and then supernatants were aliquoted into 1.5-mL tubes and centrifuged again at 1000 rpm for 5 min. Finally, all samples were aliquoted into 1.5-mL tubes and centrifuged for 5 min at 8000 rpm to collect the bacteria. Bacterial pellets were frozen in liquid nitrogen and stored at −80°C until required for RNA extraction.
RNA extraction
The bacterial RNA was extracted as described previously (Toledo-Arana et al. 2009), with slight modifications. Bacterial pellets were resuspended into 450 μL lysozyme (Sigma) at a final concentration of 15 mg/mL and incubated at 30°C for 5 min. Resuspended cells were transferred into tubes containing glass beads (Sigma) and then vortexed for 2 min. TRIZOL (1 mL) was added to the tubes, which were mixed gently and incubated for 5 min. Chloroform (100 μL) was added, and tubes were mixed gently and incubated for 3 min at room temperature. After centrifuging at 13,000 rpm at 4°C for 15 min, the upper aqueous phase was transferred into a 2-mL tube containing 200 μL chloroform, mixed and incubated for 5 min at room temperature. The tubes were centrifuged at 13,000 rpm at 4°C for 15 min. The upper aqueous phase was transferred into a 2-mL tube containing 800 μL isopropanol. The tubes were mixed, and incubated for 20 min at room temperature. After centrifuging at 13,000 rpm at 4°C for 20 min, the pellets were collected, washed with 75% ethanol, and incubated at room temperature for 3 min. The tubes were centrifuged at 13,000 rpm at 4°C for 15 min. The dried RNA pellets were then dissolved in nuclease-free water. Residual genomic DNA was removed by DNase I (New England Biolabs). RNA concentration and integrity was assessed by an Agilent 2100 Bioanalyzer (Agilent Technologies). RNAs were stored at −80°C until needed.
Construction of cDNA libraries and dRNA-seq
Libraries for strand-specific sequencing (RNA-seq) were constructed as follows: 5 μg total RNA were treated with DNase I (New England Biolabs) to remove residual genomic DNA, and rRNA was depleted by the Ribo-Zero Gram-Positive rRNA Removal Kit (Epicentre). The rRNA-depleted RNA was interrupted into an ∼150-nt fragment by high salt and high temperature. The fragment RNA was reverse-transcribed into cDNA first strand by random primers using the SuperScript II Reverse-Transcription Kit (Invitrogen) and purified by AMPure XP beads (Beckman) to remove the common dNTPs. The cDNA second strand was synthesized by the addition of dNTPs with dUTP instead of dTTP, RNase H, and DNA polymerase and incubation at 16°C for 2 h, followed by end repair and sequencing adapter ligation. The adapter ligated cDNAs were digested by Uracil N-Glycosylase (Applied Biosystems) to remove the second strand with dUTP. PCR was performed to amplify the digested cDNA, and the PCR product was purified using AMPure XP beads. The size of the purified library was 200–400 bp, and the libraries were sequenced by Illumina HiSeq 2000 paired-end 90-bp sequencing.
For TSS sequencing (TSS-seq), total RNA (5 μg) was first treated with DNase I. The DNase-treated RNA was digested by TEX (Epicentre) at 30°C for 1 h to enrich the unprocessed transcripts. The TEX-treated RNA was extracted by a phenol/chloroform/isoamyl alcohol (25:24:1, v/v) mixture to remove the exonuclease, and precipitated with ethanol. The enriched transcripts were then treated with Tobacco Acid Pyrophosphatase (TAP; Epicentre) at 37°C for 1 h to remove the 5′-triphosphates group and generate 5′-monophosphates, and again purified by organic extraction and precipitation as above. 5′ RNA adapters were ligased to the purified 5′-monophosphates RNAs by T4 RNA ligase 1 (NEB), and then the RNAs were reverse-transcribed by semirandom primers using a SuperScript II Kit (Invitrogen). The cDNA first strand was purified using Agencourt AMPure XP beads (Beckman), and the second strand was amplified by PCR using the Illumina sequencing primers and Platinum Pfx DNA Polymerase (Invitrogen). The PCR products were purified twice by Agencourt AMPure XP beads. The TSS-seq libraries were sequenced by Illumina HiSeq 2000 single-end 50-bp sequencing.
Sequence mapping
For mapping of reads from TSS-seq, 5′ end-linker sequences were clipped, and reads with a poly(A) content >70% were discarded. The remaining reads were aligned to the genome sequence of SS strain P1/7 (GenBank, Accession No. AM946016) using the segemehl program (parameter settings E 5 A 85 D 1 H 1) (Hoffmann et al. 2009). Reads that mapped with an identity of ≥85% and a minimum length of 12 nt were analyzed further, whereas reads mapping to rRNA or tRNA genes were excluded. Disregarding cDNAs <12 bp, 86.0%–89.1% of the cDNAs in each library could be mapped to the SS chromosome P1/7 (Supplemental Table S11).
For the mapping of reads from RNA-seq, SOAPaligner (version 2.20) was used (parameters: −m0 −x1000 −s40 −l 35 −v3 −r2). In each library, 69.4%–92.7% of the cDNAs could be mapped to the SS P1/7 chromosome (Supplemental Table S11).
TSS and operon annotation
A TSS was defined as the genomic position where the observed normalized number of reads at this position from the TSS-seq library exceeded that from the RNA-seq library. Because TSS-seq reads are mapped to the 5′ flank over the cDNA sequence of a gene, and RNA-seq reads are evenly distributed over the corresponding gene, we could not simply compare the TSS locus depth of the two libraries. We, therefore, derived a method to calculate the normalized depth of the two different libraries, where Dt represented the depth of the TSS-seq library, and Dr represented the depth of the RNA-seq library. Dt and Dr were calculated by the following formula:
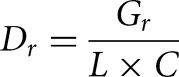 |
where Gr is the total number of nucleotides mapped to genes of the RNA-seq library, L is the average length of genes, and C is the number of genes. Gr and L were calculated by the following formula:
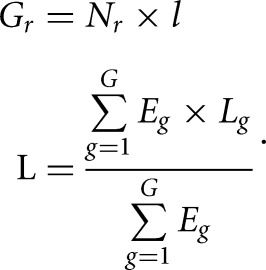 |
Nr is read number of the RNA-seq library, l is read length of the RNA-seq library, Eg is expression of the gene (g), Lg is the length of the gene, and G is the total number of genes.
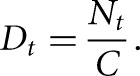 |
Nt is the total number of reads mapped to genes of the TSS library, and C is the number of genes.
We filtered TSS with parameters as follows: (1) filter TSS and genes of rRNA and tRNA; (2) filter TSS that mapped reads <10 reads; (3) filter average depth coverage of genes in RNA-seq library <10 reads; (4) filter TSS depth/(average depth of 50 bp downstream) <Dt/Dr. The TSSs were divided into five categories, according to a previous report (Sharma et al. 2010): (1) primary TSS, having the most reads within ≤500 bp upstream of an annotated ORF; (2) secondary TSS, referring to the same ORF as the primary TSS, but with fewer reads; (3) internal TSS, within an annotated ORF on the same strand; (4) antisense TSS (for identifying asRNA), located antisense to an annotated gene or within ≤50 bp of its 5′ or 3′ UTRs; and (5) orphan TSS (for identifying sRNA), with no annotated genes near the region.
To build an operon map for SS strain P1/7, three data sets were combined: (1) TSS-seq data revealing TSS; (2) conventional RNA-seq, i.e., strand-specific Solexa sequencing of cDNA to identify cotranscribed genes; and (3) 431 putative SS strain P1/7 operons predicted by the DOOR (Mao et al. 2009). The operons were assigned as one of the following classifications, according to a previous report (Sharma et al. 2010): (1) “confirmed”: an operon predicted by the DOOR was supported by dRNA-seq data; (2) “decreased”: an operon predicted by the DOOR was decreased, i.e., the dRNA-seq data indicated a shorter polycistronic mRNA than predicted; (3) “extended”: an operon predicted by the DOOR was extended, i.e., the dRNA-seq data indicated a longer polycistronic mRNA than predicted; (4) “new”: an operon was based on sequencing data and was not previously predicted by the DOOR; and (5) “alternative”: a suboperon of a confirmed, extended, or new operon, based on the presence of an intra-operonic TSS.
Prediction of promoter motifs and small ORFs
Promoter regions (50 nt upstream of the annotated TSS) were scanned with MEME (Bailey and Elkan 1994) for regulatory motifs. The detected regulatory RNA candidates could also code for short peptides; therefore, a pipeline was constructed to search for ORFs within the sRNA candidates. To identify short conserved protein-coding genes in SS, homolog sequences of SS sRNA genes in 45 Streptococcus bacterial genomes (Supplemental Table S2) were aligned with muscle (Edgar 2004). The alignments were analyzed for potential coding segments using RNAcode (Washietl et al. 2011) and a P-value cut-off of 0.05. High scoring segments were combined if they were ≤15 nt apart and in the same reading frame. The combined regions were inspected for potential ORFs starting with a canonical start codon and ending with a canonical stop codon. If no complete ORF was detected, the RNAcode high scoring segment was extended by 51 nt up- and downstream, and the analysis was repeated.
Semiquantitative analysis of expression differences
An alignment package, SOAPaligner (version 2.20), was used to map reads from RNA-seq back to the genes of SS at the parameters of “−m0 −x1000 −s40 −l 35 −v3 −r2.” The meaning and selection principles of the parameters are available at: http://soap.genomics.org.cn/soapaligner.html. Next, the number of mapped clean reads (reads mapped to rRNA sequences were also discarded) for each gene was counted and then normalized to the RPKM value (reads per kb per million reads), which is widely used to calculate the expression of genes (Mortazavi et al. 2008). A P-value was used to identify genes expressed differentially between each sample using the formula below. N1 represented the total number of clean reads in sample 1, whereas the total number of clean reads in sample 2 was noted as N2, and gene A contained x and y clean reads mapped to samples 1 and 2, respectively (Audic and Claverie 1997).
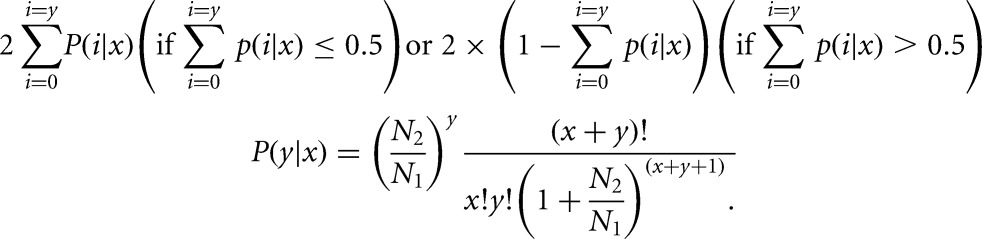 |
A false discovery rate (FDR) was applied to identify the threshold of the P-value, as used in multiple tests and analyses (Benjamini et al. 2001), and this value was calculated via SAS (version 9.1.3). In the present work, the differentially expressed genes between each of the two samples were screened with the threshold of FDR < 0.001 and the absolute value of log2 ratio ≥ 1 (Wu et al. 2010a).
Homology analysis
To identify homologs of sRNA genes in Streptococcus species, the semiglobal alignment tool Gotohscan (Hertel et al. 2009) was used with an E-value cut-off of 1 × 10−5. Homology searches were based on scans of the bacterial NCBI genome database (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/; downloaded on 09/03/2012) (Supplemental Table S2). Sequence-based alignment methods such as Gotohscan are appropriate for structural RNAs when the pairwise sequence identity is at least 50%–60% (Gardner et al. 2005). Therefore, to reduce the number of false positives, sequences identified as homologs were rejected when the sequence identity to the query sequence was <60%. Results were aligned with RNAclust (Will et al. 2007), which is based on the LocARNA algorithm (Buttner et al. 2002), and visualized with the SoupViewer (http://www.bioinf.uni-leipzig.de/software.html). To scan the SS strain P1/7 genome for known sRNAs, the Rfam database (version 11.0) was used. The annotation results are available via the Rfam website (http://rfam.sanger.ac.uk/genome/218494#tabview=tab1).
Computational prediction of sRNA targets
To predict rss03 and rss06 targets, IntaRNA was used with the following settings: −p 7 −w 140 −L 70 (Busch et al. 2008). Putative interactions were searched in the full 5′ UTR and CDS of all genes.
Construction of deletion mutants and complemented strains
To construct the knockout mutants of rss03, rss04, rss05, rss06, rss11, and rss12, upstream and downstream regions of each gene were amplified (specific primers with restriction enzyme sites are listed in Supplemental Table S12) and fused by overlap-extension PCR. The fusion fragments were purified, digested with respective endonucleases, and then cloned into the same sites of the temperature-sensitive SS-E. coli shuttle vector pSET4S (Takamatsu et al. 2001b), yielding knockout vectors. P1/7 competent cells were prepared and subjected to electrotransformation with the knockout vectors, based on a previously described method (Takamatsu et al. 2001a). Procedures for selecting mutants by allelic exchange were carried out as described previously (Takamatsu et al. 2001b), with slight modifications. Briefly, the single-crossover mutants were obtained by culturing on THA with spectinomycin at 37°C, and the double-crossover mutants were generated by repeated passaging at 28°C on THA without spectinomycin. The deletion of target genes was verified by PCR and sequencing.
The complementation of target genes into chromosomes prevents heterogeneity of gene expression due to variation in the plasmid copy number (Dortet et al. 2011). From our RNA-seq experiments, there is no transcript at the genomic intergenic region between SSU1329 and SSU1330 into which the target genes could be inserted. The complemented strains for rss03, rss04, rss05, rss06, and rss11 were constructed according to the methods of gene deletion, with some modifications. A fragment of the P1/7 genome between SSU01329 and SSU1330 was fused with the target gene and its promoter by overlap-extension PCR (Supplemental Table S12). The fusion products were cloned into pSET4S and then electroporated into competent cells of deletion mutants. The single-crossover mutants were selected as complemented strains at 37°C on THB agar under selection pressure with spectinomycin resistance. Complementation was verified by PCR and sequencing.
Northern blot analysis
Northern blot analysis was performed using the DIG Northern Starter Kit (Roche), according to the manufacturer's instructions. Briefly, total RNA (1–5 µg) was mixed with two volumes of formaldehyde loading buffer, followed by denaturation at 65°C for 10 min. Samples were electrophoresed on 2% formaldehyde-denaturing agarose gels in MOPS running buffer, and then transferred to a positively charged nylon membrane. Membranes were subsequently UV-crosslinked and probed with RNA probes. RNA probes were synthesized and labeled using the DIG RNA Labeling Kit (Roche) with PCR-generated templates, according to the manufacturer's instructions (the primers for the RNA probes are listed in Supplemental Table S12). Membranes were prehybridized for 30 min in DIG Easy Hyb, and hybridizations were performed overnight at 68°C. After hybridization and stringency washes, the membranes were further washed with washing buffer and blocking buffer from the kit. The membranes were incubated with antibody solution at room temperature for 30 min. After the membranes were washed in washing buffer and equilibrated in detection buffer, CDP-Star was added to the membranes. The membranes were exposed to X-ray film.
Assay for bacteria survival in pig blood
Susceptibility assays were performed as previously described (Echenique-Rivera et al. 2011). P1/7 wild-type and deletion mutant strains were cultured to the exponential growth phase (OD600 nm ∼0.6). Approximately 108 bacteria were pelleted by centrifugation at 8000 rpm for 5 min, washed twice in PBS, and then resuspended in an equal volume (1 mL) of freshly isolated pig blood. Mixtures were incubated at 37°C with occasional gentle shaking to avoid sedimentation. An aliquot of the infected whole blood cultures was removed at 0 h and 4 h, and the number of viable bacteria was determined by plating serial dilutions onto THA and incubating overnight at 37°C. The survival results were expressed as a fold change relative to the level of wild-type strain P1/7. Statistical analyses were performed using one-way ANOVA and Dunnett's multiple comparisons test. Experiments were performed in triplicate.
Challenge with P1/7 wild-type strain and mutants in zebrafish
P1/7 wild-type strain, mutants, and complemented strains were grown to exponential phase and collected, washed twice in PBS, and adjusted to the appropriate doses. Zebrafish were anesthetized with tricaine methanesulfonate (MS-222; Hangzhou Animal Medicine Factory) at a concentration of 90 mg/L. Fifteen zebrafish per dose were intraperitoneally injected with 10-fold serially diluted suspensions containing 104–108 CFU/fish of bacteria in PBS. Negative controls were injected with PBS. Bacterial CFUs of injected inoculum were confirmed by plating on THA. Mortality was monitored until 7 d post-infection. The experiments were repeated three times. The 50% lethal dose (LD50) results were calculated using the method by Reed and Muench. An unpaired t-test was used to compare log10LD50 between strains P1/7 and Δrss12. For the remaining comparisons, statistical analyses were performed using one-way ANOVA and Dunnett's multiple comparisons test. (*) P< 0.05 and (**) P < 0.01.
qRT-PCR analysis
For qRT-PCR analysis, bacterial RNA (1 µg) was reverse-transcribed to cDNA using a PrimeScript reagent kit (Takara), according to the manufacturer's instructions. Primers for qRT-PCR are listed in Supplemental Table S12. The qRT-PCR analysis was performed using a 7300 Real-Time PCR System (ABI) with SYBR Premix Ex Taq (Takara), according to the manufacturer's instructions. Each sample was run in triplicate. Housekeeping gene parC, which encodes DNA topoisomerase IV subunit A, was used as the internal control, and was also amplified under the same conditions. After PCR amplification was complete, the relative fold change after stimulation was calculated based on the 2−ΔΔCT method (Livak and Schmittgen 2001). Statistical analyses were performed using one-way ANOVA and Dunnett's multiple comparisons test.
Simultaneous mapping of 5′ and 3′ ends of RNA molecules
The simultaneous mapping of 5′ and 3′ RNA ends by RACE was performed as previously described (Toledo-Arana et al. 2009), with the following modifications. Total RNA (10 μg) was treated with DNase I (New England Biolabs), which was then inactivated by adding 1 μL 0.1 M EDTA and heating at 75°C for 10 min. After ethanol precipitation purification, the RNAs were divided into two aliquots. Both aliquots were incubated for 45 min at 37°C with 1× TAP Buffer, in the presence or absence of 10 units TAP (Epicentre). The RNAs were then extracted with phenol/chloroform/isoamyl alcohol and precipitated with ethanol. The purified TAP+ and TAP− treated RNAs (500 ng in 10 μL RNase-free water) were denatured by heating for 5 min at 95°C and chilling on ice for 5 min. The denatured RNAs were ligated with 40 units of T4 RNA Ligase I in the presence of 1× RNA Ligase I Buffer, 8% DMSO, 10 units of RNase Inhibitor, 1 unit of DNase I and RNase-free ddH2O in a total volume of 25 µL at 17°C overnight. After purification as described above, the circularized RNAs were dissolved in 10 µL RNase-free water. Reverse-transcription (RT) was performed using primer A (Supplemental Table S12) and the SuperScript II Reverse Transcriptase Kit (Invitrogen). The RT products were added to corresponding primer B and amplified by PCR with Taq enzyme (Takara). The RT-PCR products were loaded on a 2% agarose gel (or 6% PAGE gel) to check for the specific band sizes, and the band present in the TAP+ reaction was recovered by the QIAquick Gel Extraction Kit (Qiagen). The purified fragments were cloned using the TOPO TA Cloning Kit (Invitrogen). Plasmids containing the expected size of insert were sent to sequencing. Sequences were compared with the SS P1/7 genome to determine the localization of the 5′ and 3′ ends and, subsequently, the sRNA sizes.
Quantification of CPS level by Dot-blot
Dot-blot analysis was used to quantify the level of CPS produced by SS under the different experimental conditions, according to previous reports (Rubens et al. 1987; Chaffin et al. 2000, 2005). Before performing Dot-blot, the anti-serotype 2 polyclonal antibody (Wang et al. 2012, 2013) was absorbed by the P1/7 CPS-deficient mutant (obtained by transposon mutagenesis) (C Ju and C Lu, unpubl.), according to previous reports (Mittal et al. 1982; Higgins and Gottschalk 1990). The absorbed serum was considered as a specific antibody against SS2 CPS. Incubation of bacteria in pig blood or CSF was performed as described above. Aliquots (3 μL) of twofold serially diluted bacteria in PBS were spotted onto a nitrocellulose membrane, fixed for 5 min with 70% ethanol, air-dried, and blocked with blocking solution (5% w/v skim milk in PBS containing 0.05% Tween 20) for 2 h. Development of the membrane essentially followed the protocol of the Pierce ECL Western Blotting Kit (Thermo Scientific), using the absorbed serum specific against SS2 CPS (dilution 1:500) as a primary antibody and HRP-conjugated anti-rabbit (Boster; dilution 1:2000) as a secondary antibody. The Dot-blot signals were then detected by a FUJIFILF LAS-4000 image analyzer. Under the same experimental conditions, the P1/7 CPS-deficient mutant incubated in THB was also spotted as a control.
RT-PCR
RT-PCRs were performed to validate operons 108 and 109 and to determine the presence of sRNA in wild-type strain P1/7, sRNA deletion mutants, and sRNAs complemented strains. RT-PCR was performed according to the protocol of the HiScript 1st Strand cDNA Synthesis Kit (Vazyme). Specific primers for RT-PCR are listed in Supplemental Table S12.
Site-directed mutagenesis
To verify whether sRNAs rss03 and rss06 interact directly with SSU0516 and SSU0308 transcripts, respectively, base substitutions were introduced into the predicted pairing region of rss03 (rss03-m3) and rss06 (rss06-m6) using a Quick Change Lightning Site-Directed Mutagenesis Kit (Agilent Technologies), according to the manufacturer's instructions. The base substitutions were generated based on plasmids pSET4S-03C and pSET4S-06C. The plasmids pSET4S-03Cm3 containing rss03-m3 and pSET4S-06Cm6 containing rss06-m6 were verified by sequencing. The protocol for complementing rss03-m3 and rss06-m6 into the Δrss03 or Δrss06 competent cells, respectively, was performed as described above.
Analysis of sRNA and mRNA interactions
Plasmids used for interaction studies were obtained from Dr. Jörg Vogel (RNA Biology Group, Institute for Molecular Infection Biology, University of Würzburg, Germany). Plasmid constructions of the 0516::GFP fusion, 0308::GFP fusion, rss03 expression, and rss06 expression have been described in detail previously (Urban and Vogel 2007). The interaction studies were carried out in E. coli strain Top10. E. coli cells were grown in LB broth at 37°C in the presence of 100 μg/mL ampicillin or 25 μg/mL chloramphenicol. Putative sRNA target sequences are cloned as translational fusions to GFP on the low-copy vectors pXG-10 or pXG-30 (Urban and Vogel 2007). For the construction of the 0308::GFP fusion in pXG-10, the full-length 5′ UTR and the amino acid residues of SSU0308 (from 30 to 40) were amplified by PCR, digested, and ligated into the Mph1103I and NheI sites of the pXG-10 plasmid. The inclusion of signal sequences was avoided to prevent traffic of the fusion proteins to the periplasm/membrane (Urban and Vogel 2007). The intra-operonic fusion of SSU0516::GFP in pXG-30 was constructed as above, but the sense oligonucleotide was annealed to the C-terminal coding region of the upstream gene SSU0515. pXG-0 does not contain gfp and was, therefore, used to determine the basal fluorescence level of E. coli cells and how it was affected by sRNA overexpression. pXG-1 expresses full-length GFP and was used to determine the general effect of the plasmid-borne sRNA gene on GFP expression. sRNAs rss03 and rss06 expression plasmids were constructed based on a high-copy vector pZE12-luc. The control plasmid pJV300 was constructed as described previously (Sittka et al. 2007). The primers used for the interaction studies are listed in Supplemental Table S12. The effect of a given sRNA on a target fusion was determined by colony fluorescence and Western blot analysis.
For colony fluorescence, E .coli Top10 cells carrying both the target-GFP fusion plasmid and sRNA-expressing plasmid were streaked on standard LB plates supplemented with the appropriate antibiotics. After overnight growth, colonies were photographed in a FUJI LAS-4000 image analyzer.
For Western blotting, strains were cultured to OD600 nm = 1, pelleted by centrifugation, and resuspended in loading buffer to a final concentration of 0.01 OD/μL. A total of 0.05 OD of whole-cell protein fractions were separated by polyacrylamide gel electrophoresis on a 15% gel, before being transferred onto a PVDF membrane using a semidry blotting apparatus (GE Healthcare). The protocol of the Pierce ECL Western Blotting Kit (Thermo Scientific) was followed for developing the membrane, using a monoclonal antibody against GFP (TransGen Biotech; dilution 1:2000) or anti-groEL antibody (Abcam; dilution 1:1000) as primary antibodies and HRP-conjugated anti-mouse (Boster; dilution 1:2000) or anti-rabbit (Boster; dilution 1:2000) as secondary antibodies. The Western blot signals were detected using a FUJIFILF LAS-4000 image analyzer, and the results were analyzed by ImageJ (http://rsb.info.nih.gov/ij/).
Validation of the revised start codon for SSU0647
The 0647::GFP fusion plasmid was generated as follows: The gfp gene (starting from the second residue of GFP) from plasmid pEGFP/N2 and the SSU0647 fragment from 200 bp upstream of the annotated start codon to 33 bp downstream from the new start codon were amplified with specific primers and fused by overlap-extension PCR. The resulting fragments were digested by endonucleases and inserted into the E. coli-S. suis shuttle vector pSET4S at the PstI/BamHI sites. Transformants were screened on LB plates containing spectinomycin. Then, the “A” bases just downstream from the old and new ATG sites of SSU0647 were deleted to generate mutant vectors 0647::GFP-m1 and 0647::GFP-m2, respectively. This was carried out by site-directed mutagenesis using the Quick Change Lightning Site-Directed Mutagenesis Kit (Agilent Technologies), according to the manufacturer's instructions. After the fusion plasmids were electrotransformed into SS strain P1/7, the strain harboring the vector 0647::GFP or derived mutant vectors were plated on THA with spectinomycin at 28°C. Western blotting of SS P1/7 harboring pSET4s, 0647::GFP, 0647::GFP-m1, and 0647::GFP-m2 fusion plasmids was performed as described above using anti-GFP (TransGen Biotech) as the primary antibody and HRP-conjugated anti-mouse as the secondary antibody (Boster).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Prof. Pascale Cossart, Dr. Nina Sesto, Dr. Pascale Romby, and Prof. Xiang Mao for their helpful discussions. We thank Prof. Jörg Vogel for providing the complete pXG plasmid system. This work was supported by grants from the Fundamental Research Funds for the Central Universities (KYZ201314), Chinese National Programs for Fundamental Research and Development (2012CB518804), the Youth Foundation of National Natural Science Foundation of China (31101828), the Natural Science Foundation of Jiangsu Province of China (No. BK2011644), the Special Fund for Public Welfare Industry of Chinese MoA (No. 201303041), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.041822.113.
REFERENCES
- Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38: 868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda J, Garrido ME, Cortes P, Llagostera M, Barbe J 2008. Analysis of the protective capacity of three Streptococcus suis proteins induced under divalent-cation-limited conditions. Infect Immun 76: 1590–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda J, Cortes P, Garrido ME, Fittipaldi N, Llagostera M, Gottschalk M, Barbe J 2009a. Contribution of the FeoB transporter to Streptococcus suis virulence. Int Microbiol 12: 137–143 [PubMed] [Google Scholar]
- Aranda J, Garrido ME, Fittipaldi N, Cortes P, Llagostera M, Gottschalk M, Barbe J 2009b. Protective capacities of cell surface-associated proteins of Streptococcus suis mutants deficient in divalent cation-uptake regulators. Microbiology 155: 1580–1587 [DOI] [PubMed] [Google Scholar]
- Aranda J, Teixido L, Fittipaldi N, Cortes P, Llagostera M, Gottschalk M, Barbe J 2012. Inactivation of the gene encoding zinc-binding lipoprotein 103 impairs the infectivity of Streptococcus suis. Can J Vet Res 76: 72–76 [PMC free article] [PubMed] [Google Scholar]
- Audic S, Claverie JM 1997. The significance of digital gene expression profiles. Genome Res 7: 986–995 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I 2001. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125: 279–284 [DOI] [PubMed] [Google Scholar]
- Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. 2007. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 21: 1353–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifait L, de la Cruz Dominguez-Punaro M, Vaillancourt K, Bart C, Slater J, Frenette M, Gottschalk M, Grenier D 2010. The cell envelope subtilisin-like proteinase is a virulence determinant for Streptococcus suis. BMC Microbiol 10: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A 2012. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41: D226–D232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Richter AS, Backofen R 2008. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 24: 2849–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner D, Nennstiel D, Klusener B, Bonas U 2002. Functional analysis of HrpF, a putative type III translocon protein from Xanthomonas campestris pv. vesicatoria. J Bacteriol 184: 2389–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Feng Y, Wang C, Zheng F, Li M, Liao H, Mao Y, Pan X, Wang J, Hu D, et al. 2011. Functional definition of LuxS, an autoinducer-2 (AI-2) synthase and its role in full virulence of Streptococcus suis serotype 2. J Microbiol 49: 1000–1011 [DOI] [PubMed] [Google Scholar]
- Chaffin DO, Beres SB, Yim HH, Rubens CE 2000. The serotype of type Ia and III group B streptococci is determined by the polymerase gene within the polycistronic capsule operon. J Bacteriol 182: 4466–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffin DO, Mentele LM, Rubens CE 2005. Sialylation of group B streptococcal capsular polysaccharide is mediated by cpsK and is required for optimal capsule polymerization and expression. J Bacteriol 187: 4615–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier C, Boisset S, Romilly C, Masquida B, Fechter P, Geissmann T, Vandenesch F, Romby P 2010. Staphylococcus aureus RNAIII binds to two distant regions of coa mRNA to arrest translation and promote mRNA degradation. PLoS Pathog 6: e1000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471: 602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Punaro MC, Segura M, Plante MM, Lacouture S, Rivest S, Gottschalk M 2007. Streptococcus suis serotype 2, an important swine and human pathogen, induces strong systemic and cerebral inflammatory responses in a mouse model of infection. J Immunol 179: 1842–1854 [DOI] [PubMed] [Google Scholar]
- Dominguez-Punaro Mde L, Segura M, Contreras I, Lachance C, Houde M, Lecours MP, Olivier M, Gottschalk M 2010. In vitro characterization of the microglial inflammatory response to Streptococcus suis, an important emerging zoonotic agent of meningitis. Infect Immun 78: 5074–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori MA, Wiemer EA, Dussurget O, Cossart P 2011. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog 7: e1002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echenique-Rivera H, Muzzi A, Del Tordello E, Seib KL, Francois P, Rappuoli R, Pizza M, Serruto D 2011. Transcriptome analysis of Neisseria meningitidis in human whole blood and mutagenesis studies identify virulence factors involved in blood survival. PLoS Pathog 7: e1002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyraud A, Tattevin P, Chabelskaya S, Felden B 2014. A small RNA controls a protein regulator involved in antibiotic resistance in Staphylococcus aureus. Nucleic Acids Res 10.1093/nar/gku149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Cao M, Shi J, Zhang H, Hu D, Zhu J, Zhang X, Geng M, Zheng F, Pan X, et al. 2012. Attenuation of Streptococcus suis virulence by the alteration of bacterial surface architecture. Sci Rep 2: 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fittipaldi N, Collis T, Prothero B, Gottschalk M 2009. Streptococcus suis meningitis, Hawaii. Emerg Infect Dis 15: 2067–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fittipaldi N, Segura M, Grenier D, Gottschalk M 2012. Virulence factors involved in the pathogenesis of the infection caused by the swine pathogen and zoonotic agent Streptococcus suis. Future Microbiol 7: 259–279 [DOI] [PubMed] [Google Scholar]
- Fulde M, Willenborg J, de Greeff A, Benga L, Smith HE, Valentin-Weigand P, Goethe R 2011. ArgR is an essential local transcriptional regulator of the arcABC operon in Streptococcus suis and is crucial for biological fitness in an acidic environment. Microbiology 157: 572–582 [DOI] [PubMed] [Google Scholar]
- Gardner PP, Wilm A, Washietl S 2005. A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Res 33: 2433–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk M, Segura M 2000. The pathogenesis of the meningitis caused by Streptococcus suis: the unresolved questions. Vet Microbiol 76: 259–272 [DOI] [PubMed] [Google Scholar]
- Gottschalk M, Xu J, Calzas C, Segura M 2010. Streptococcus suis: a new emerging or an old neglected zoonotic pathogen? Future Microbiol 5: 371–391 [DOI] [PubMed] [Google Scholar]
- Han H, Liu C, Wang Q, Xuan C, Zheng B, Tang J, Yan J, Zhang J, Li M, Cheng H, et al. 2012. The two-component system Ihk/Irr contributes to the virulence of Streptococcus suis serotype 2 strain 05ZYH33 through alteration of the bacterial cell metabolism. Microbiology 158: 1852–1866 [DOI] [PubMed] [Google Scholar]
- Hertel J, de Jong D, Marz M, Rose D, Tafer H, Tanzer A, Schierwater B, Stadler PF 2009. Non-coding RNA annotation of the genome of Trichoplax adhaerens. Nucleic Acids Res 37: 1602–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins R, Gottschalk M 1990. An update on Streptococcus suis identification. J Vet Diagn Invest 2: 249–252 [DOI] [PubMed] [Google Scholar]
- Hill JE, Gottschalk M, Brousseau R, Harel J, Hemmingsen SM, Goh SH 2005. Biochemical analysis, cpn60 and 16S rDNA sequence data indicate that Streptococcus suis serotypes 32 and 34, isolated from pigs, are Streptococcus orisratti. Vet Microbiol 107: 63–69 [DOI] [PubMed] [Google Scholar]
- Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermuller J 2009. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 5: e1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden MT, Hauser H, Sanders M, Ngo TH, Cherevach I, Cronin A, Goodhead I, Mungall K, Quail MA, Price C, et al. 2009. Rapid evolution of virulence and drug resistance in the emerging zoonotic pathogen Streptococcus suis. PLoS One 4: e6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Boisset S, Saveanu C, Benito Y, Geissmann T, Namane A, Lina G, Etienne J, Ehresmann B, Ehresmann C, et al. 2005. Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J 24: 824–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju CX, Gu HW, Lu CP 2012. Characterization and functional analysis of atl, a novel gene encoding autolysin in Streptococcus suis. J Bacteriol 194: 1464–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Shah P, Swiatlo E, Burgess SC, Lawrence ML, Nanduri B 2010. Identification of novel non-coding small RNAs from Streptococcus pneumoniae TIGR4 using high-resolution genome tiling arrays. BMC Genomics 11: 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecours MP, Gottschalk M, Houde M, Lemire P, Fittipaldi N, Segura M 2011. Critical role for Streptococcus suis cell wall modifications and suilysin in resistance to complement-dependent killing by dendritic cells. J Infect Dis 204: 919–929 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Hong SH 2012. Analysis of microRNA-size, small RNAs in Streptococcus mutans by deep sequencing. FEMS Microbiol Lett 326: 131–136 [DOI] [PubMed] [Google Scholar]
- Le Rhun A, Charpentier E 2012. Small RNAs in streptococci. RNA Biol 9: 414–426 [DOI] [PubMed] [Google Scholar]
- Li M, Wang C, Feng Y, Pan X, Cheng G, Wang J, Ge J, Zheng F, Cao M, Dong Y, et al. 2008. SalK/SalR, a two-component signal transduction system, is essential for full virulence of highly invasive Streptococcus suis serotype 2. PLoS One 3: e2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tan C, Zhou Y, Fu S, Hu L, Hu J, Chen H, Bei W 2011. The two-component regulatory system CiaRH contributes to the virulence of Streptococcus suis 2. Vet Microbiol 148: 99–104 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Loh E, Gripenland J, Johansson J 2006. Control of Listeria monocytogenes virulence by 5′-untranslated RNA. Trends Microbiol 14: 294–298 [DOI] [PubMed] [Google Scholar]
- Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J 2009. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139: 770–779 [DOI] [PubMed] [Google Scholar]
- Mann B, van Opijnen T, Wang J, Obert C, Wang YD, Carter R, McGoldrick DJ, Ridout G, Camilli A, Tuomanen EI, et al. 2012. Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog 8: e1002788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao F, Dam P, Chou J, Olman V, Xu Y 2009. DOOR: a database for prokaryotic operons. Nucleic Acids Res 37: D459–D463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschke J, Georg J, Scholz I, Sharma CM, Dienst D, Bantscheff J, Voss B, Steglich C, Wilde A, Vogel J, et al. 2010. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci 108: 2124–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal KR, Higgins R, Lariviere S 1982. Evaluation of slide agglutination and ring precipitation tests for capsular serotyping of Haemophilus pleuropneumoniae. J Clin Microbiol 15: 1019–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Pan X, Ge J, Li M, Wu B, Wang C, Wang J, Feng Y, Yin Z, Zheng F, Cheng G, et al. 2009. The orphan response regulator CovR: a globally negative modulator of virulence in Streptococcus suis serotype 2. J Bacteriol 191: 2601–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon C, du Merle L, Caliot ME, Trieu-Cuot P, Le Bouguenec C 2012. An in silico model for identification of small RNAs in whole bacterial genomes: characterization of antisense RNAs in pathogenic Escherichia coli and Streptococcus agalactiae strains. Nucleic Acids Res 40: 2846–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R 2004. Fast and effective prediction of microRNA/target duplexes. RNA 10: 1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter AS, Schleberger C, Backofen R, Steglich C 2010. Seed-based INTARNA prediction combined with GFP-reporter system identifies mRNA targets of the small RNA Yfr1. Bioinformatics 26: 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romby P, Vandenesch F, Wagner EG 2006. The role of RNAs in the regulation of virulence-gene expression. Curr Opin Microbiol 9: 229–236 [DOI] [PubMed] [Google Scholar]
- Rubens CE, Wessels MR, Heggen LM, Kasper DL 1987. Transposon mutagenesis of type III group B Streptococcus: correlation of capsule expression with virulence. Proc Natl Acad Sci 84: 7208–7212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheld WM, Whitley RJ, Marra CM 2004. Infections of the central nervous system. Lippincott, PA [Google Scholar]
- Schmidtke C, Findeiss S, Sharma CM, Kuhfuss J, Hoffmann S, Vogel J, Stadler PF, Bonas U 2012. Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions. Nucleic Acids Res 40: 2020–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider O, Michel U, Zysk G, Dubuis O, Nau R 1999. Clinical outcome in pneumococcal meningitis correlates with CSF lipoteichoic acid concentrations. Neurology 53: 1584–1587 [DOI] [PubMed] [Google Scholar]
- Seele J, Singpiel A, Spoerry C, von Pawel-Rammingen U, Valentin-Weigand P, Baums CG 2013. Identification of a novel host-specific IgM protease in Streptococcus suis. J Bacteriol 195: 930–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma CM, Darfeuille F, Plantinga TH, Vogel J 2007. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev 21: 2804–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, et al. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464: 250–255 [DOI] [PubMed] [Google Scholar]
- Sittka A, Pfeiffer V, Tedin K, Vogel J 2007. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol Microbiol 63: 193–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small PM, Tauber MG, Hackbarth CJ, Sande MA 1986. Influence of body temperature on bacterial growth rates in experimental pneumococcal meningitis in rabbits. Infect Immun 52: 484–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HE, Vecht U, Wisselink HJ, Stockhofe-Zurwieden N, Biermann Y, Smits MA 1996. Mutants of Streptococcus suis types 1 and 2 impaired in expression of muramidase-released protein and extracellular protein induce disease in newborn germfree pigs. Infect Immun 64: 4409–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smollett KL, Fivian-Hughes AS, Smith JE, Chang A, Rao T, Davis EO 2009. Experimental determination of translational start sites resolves uncertainties in genomic open reading frame predictions – application to Mycobacterium tuberculosis. Microbiology 155: 186–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu D, Osaki M, Sekizaki T 2001a. Construction and characterization of Streptococcus suis-Escherichia coli shuttle cloning vectors. Plasmid 45: 101–113 [DOI] [PubMed] [Google Scholar]
- Takamatsu D, Osaki M, Sekizaki T 2001b. Thermosensitive suicide vectors for gene replacement in Streptococcus suis. Plasmid 46: 140–148 [DOI] [PubMed] [Google Scholar]
- Tang F, Zhang W, Lu C 2013. Lysogenic Streptococcus suis isolate SS2-4 containing prophage SMP showed increased mortality in zebra fish compared to the wild-type isolate. PLoS One 8: e54227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesorero RA, Yu N, Wright JO, Svencionis JP, Cheng Q, Kim JH, Cho KH 2013. Novel regulatory small RNAs in Streptococcus pyogenes. PLoS One 8: e64021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, et al. 2009. The Listeria transcriptional landscape from saprophytism to virulence. Nature 459: 950–956 [DOI] [PubMed] [Google Scholar]
- Tuomanen E, Tomasz A, Hengstler B, Zak O 1985. The relative role of bacterial cell wall and capsule in the induction of inflammation in pneumococcal meningitis. J Infect Dis 151: 535–540 [DOI] [PubMed] [Google Scholar]
- Urban JH, Vogel J 2007. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res 35: 1018–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier M, Geelen SP, Kimpen JL, Hoepelman IM, Tuomanen EI 2003. Reprogramming the host response in bacterial meningitis: how best to improve outcome? Clin Microbiol Rev 16: 415–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderpool CK, Balasubramanian D, Lloyd CR 2011. Dual-function RNA regulators in bacteria. Biochimie 93: 1943–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Li M, Feng Y, Zheng F, Dong Y, Pan X, Cheng G, Dong R, Hu D, Feng X, et al. 2009. The involvement of sortase A in high virulence of STSS-causing Streptococcus suis serotype 2. Arch Microbiol 191: 23–33 [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang W, Wu Z, Zhu X, Lu C 2011. Functional analysis of luxS in Streptococcus suis reveals a key role in biofilm formation and virulence. Vet Microbiol 152: 151–160 [DOI] [PubMed] [Google Scholar]
- Wang K, Sun X, Lu C 2012. Development of rapid serotype-specific PCR assays for eight serotypes of Streptococcus suis. J Clin Microbiol 50: 3329–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Zhang W, Li X, Lu C, Chen J, Fan W, Huang B 2013. Characterization of Streptococcus suis isolates from slaughter swine. Curr Microbiol 66: 344–349 [DOI] [PubMed] [Google Scholar]
- Washietl S, Findeiss S, Muller SA, Kalkhof S, von Bergen M, Hofacker IL, Stadler PF, Goldman N 2011. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA 17: 578–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Storz G 2009. Regulatory RNAs in bacteria. Cell 136: 615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R 2007. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput Biol 3: e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenborg J, Fulde M, de Greeff A, Rohde M, Smith HE, Valentin-Weigand P, Goethe R 2011. Role of glucose and CcpA in capsule expression and virulence of Streptococcus suis. Microbiology 157: 1823–1833 [DOI] [PubMed] [Google Scholar]
- Wilson TL, Jeffers J, Rapp-Gabrielson VJ, Martin S, Klein LK, Lowery DE, Fuller TE 2007. A novel signature-tagged mutagenesis system for Streptococcus suis serotype 2. Vet Microbiol 122: 135–145 [DOI] [PubMed] [Google Scholar]
- Wu T, Chang H, Tan C, Bei W, Chen H 2009. The orphan response regulator RevSC21 controls the attachment of Streptococcus suis serotype-2 to human laryngeal epithelial cells and the expression of virulence genes. FEMS Microbiol Lett 292: 170–181 [DOI] [PubMed] [Google Scholar]
- Wu J, Zhang Y, Zhang H, Huang H, Folta KM, Lu J 2010a. Whole genome wide expression profiles of Vitis amurensis grape responding to downy mildew by using Solexa sequencing technology. BMC Plant Biol 10: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Zhang W, Lu Y, Lu C 2010b. Transcriptome profiling of zebrafish infected with Streptococcus suis. Microb Pathog 48: 178–187 [DOI] [PubMed] [Google Scholar]
- Wurtzel O, Sesto N, Mellin JR, Karunker I, Edelheit S, Becavin C, Archambaud C, Cossart P, Sorek R 2012. Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol Syst Biol 8: 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Jing H, Chen Z, Zheng H, Zhu X, Wang H, Wang S, Liu L, Zu R, Luo L, et al. 2006. Human Streptococcus suis outbreak, Sichuan, China. Emerg Infect Dis 12: 914–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Lu CP 2007. Immunoproteomic assay of membrane-associated proteins of Streptococcus suis type 2 China vaccine strain HA9801. Zoonoses Public Health 54: 253–259 [DOI] [PubMed] [Google Scholar]
- Zhang A, Mu X, Chen B, Han L, Chen H, Jin M 2011. IgA1 protease contributes to the virulence of Streptococcus suis. Vet Microbiol 148: 436–439 [DOI] [PubMed] [Google Scholar]
- Zheng F, Ji H, Cao M, Wang C, Feng Y, Li M, Pan X, Wang J, Qin Y, Hu F, et al. 2011. Contribution of the Rgg transcription regulator to metabolism and virulence of Streptococcus suis serotype 2. Infect Immun 79: 1319–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]