

Abstract
Oxidative stress in the myocardium plays an important role in the pathophysiology of hemodynamic overload. The mechanism by which reactive oxygen species (ROS) in the cardiac myocyte mediate myocardial failure in hemodynamic overload is not known. Accordingly, our goals were to test whether myocyte-specific overexpression of peroxisomal catalase (pCAT) that localizes in the sarcoplasm protects mice from hemodynamic overload-induced failure and prevents oxidation and inhibition of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), an important sarcoplasmic protein. Chronic hemodynamic overload was caused by ascending aortic constriction (AAC) for 12 wk in mice with myocyte-specific transgenic expression of pCAT. AAC caused left ventricular hypertrophy and failure associated with a generalized increase in myocardial oxidative stress and specific oxidative modifications of SERCA at cysteine 674 and tyrosine 294/5. pCAT overexpression ameliorated myocardial hypertrophy and apoptosis, decreased pathological remodeling, and prevented the progression to heart failure. Likewise, pCAT prevented oxidative modifications of SERCA and increased SERCA activity without changing SERCA expression. Thus cardiac myocyte-restricted expression of pCAT effectively ameliorated the structural and functional consequences of chronic hemodynamic overload and increased SERCA activity via a post-translational mechanism, most likely by decreasing inhibitory oxidative modifications. In pressure overload-induced heart failure cardiac myocyte cytosolic ROS play a pivotal role in mediating key pathophysiologic events including hypertrophy, apoptosis, and decreased SERCA activity.
Keywords: apoptosis, catalase, H2O2, hypertrophy, SERCA
the role of reactive oxygen species (ROS) in mediating myocardial failure in response to chronic hemodynamic overload is well appreciated (29, 32, 34). Systemic administration of small molecule antioxidants ameliorated pathological remodeling and failure in mice with hemodynamic overload due to chronic aortic constriction (8, 35). Transgenic total body overexpression of mitochondrial catalase (mCAT) likewise decreased left ventricular (LV) hypertrophy and failure in mice with aortic constriction (6), further suggesting that mitochondria are a source of ROS in hemodynamic overload. However, several nonmitochondrial sources of ROS in the myocardium have been implicated in hemodynamic overload including uncoupled endothelial nitric oxide synthase (23, 33), NADPH oxidases (4), and xanthine oxidase (21). Because these nonmitochondrial sources of ROS are located in the sarcoplasm, we reasoned that their effects should be susceptible to catalase that is targeted to the sarcoplasm of the cardiac myocyte. Accordingly, our first goal was to test whether LV hypertrophy and failure following ascending aortic constriction (AAC) is inhibited in transgenic mice in which catalase is targeted to cardiac myocyte peroxisomes (pCAT), and thus located in the cytosolic compartment.
An important consequence of increased myocyte ROS may be decreased sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) activity. In animal models of pressure overload-induced heart failure the activity of SERCA is decreased and contributes to depletion of sarcoplasmic reticulum calcium stores, decreased intracellular calcium transients, and impaired contractile function (12, 14, 15, 25, 26). The importance of impaired SERCA function in pressure overload is supported by the observation that increasing myocardial SERCA levels corrects calcium handling and myocardial function (9, 14, 22, 25), whereas SERCA+/− mice have accelerated progression to failure (31).
Decreased SERCA activity in pressure overload is often associated with decreased expression of SERCA (26). However, there is increasing recognition that SERCA activity can also be inhibited by irreversible oxidative post-translational modifications including sulfonylation at cysteine 674 and nitration at tyrosine 294/5 (1, 18, 28, 38), which might act alone or in concert with a decrease in protein expression, to limit SERCA activity. Therefore, our second goal was to test whether pCAT can prevent SERCA oxidation and rescue SERCA function in mice with pressure overload due to aortic constriction.
METHODS
Experimental animals.
Transgenic FVB/N mice with myocyte-specific overexpression of catalase in the cytosolic compartment (pCAT) (16, 20, 39) and wild-type (WT) FVB/N mice were used in this study. The protocol was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine.
AAC.
The surgical procedure was performed as we have described (17). Briefly, 10-wk-old male WT and pCAT mice were anesthetized by an intraperitoneal injection with pentobarbital (50 mg/kg), and the chest was shaved. Animals were intubated with a 20-gauge intravenous catheter, ventilated on a rodent respirator (Harvard Apparatus, Holliston, MA) with a tidal volume of 0.2 ml at a respiratory rate of 130 breaths/min. The thorax was opened by an anterolateral thoracotomy, and AAC was performed by tying a 7-0 silk suture around the ascending aorta and a 27-gauge needle, which was then promptly removed after ligation. The chest was closed and mice were allowed to recover on a warming pad until they were fully awake. Sham-operated mice underwent a similar procedure without ligation of the ascending aorta.
Echocardiographic measurements.
LV dimensions and function were measured in nonanesthetized mice shortly before surgery and 1, 2, 4, 8, and 12 wk after surgery using an Acuson Sequoia C-256 echocardiograph machine equipped with a 15-MHz linear transducer (model 15L8) as we have described (27). Briefly, the heart was imaged in the two-dimensional parasternal short-axis view, and an M-mode echocardiogram of the midventricle was recorded at the level of papillary muscles. Anterior wall thickness (AWT), posterior wall thickness (PWT), LV end-diastolic dimension (EDD) and end-systolic dimension (ESD) were measured from the M-mode image. LV fractional shortening (FS) was calculated as (EDD − ESD)/EDD × 100.
Exercise capacity.
Maximal exercise capacity was tested 12 wk after surgery by using a rodent treadmill with air puff motivation, as we have described (3). Exhaustion is defined as the point at which the animal cannot keep pace with the treadmill (within 15 s) despite air puff motivation. Maximal exercise capacity was calculated as the total distance run by the animal.
Organ weight and histology.
Mice were euthanized 12 wk after surgery. Heart, LV with septum, lung, and liver were weighed. LV samples were fixed in 10% buffered formalin, embedded with paraffin, and sectioned. To assess myocyte size, sections were stained with hematoxyline and eosin and examined under a light microscopy (BX 40; Olympus). Five random fields from each of four sections per animal were analyzed so that 60 myocytes per animal were measured. Myocyte cross-sectional area was measured using National Institutes of Health (NIH) ImageJ software as we have described (27). To assess fibrosis, sections were stained with Masson's trichrome kit (Sigma) and examined under a light microscope (BX 40; Olympus).
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling staining.
Apoptosis was assessed using an In Situ Cell Death Detection Fluorescein Kit (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions, as we have described (27). Briefly, LV sections were incubated with the reaction mixture containing terminal deoxynucleotidyl transferase and fluorescein-labeled dUTP. To identify cardiomyocytes, sections were incubated with monoclonal antibody to mouse anti-α-sarcomeric actin (Sigma-Aldrich, St. Louis, MO) and incubated with goat anti-mouse IgG conjugated TRITC (Sigma). Finally, to identify all nuclei (nonapoptotic and apoptotic), sections were stained with Hoechst 33258. Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL)-positive myocytes were identified by colocalization fragmented nuclear DNA, nuclei (Hoechst 33342), and α-sarcomeric actin. Specificity of the technique to detect DNA fragmentation was documented by positive labeling of nuclei after exposure to DNase I, and lack of labeling when terminal deoxynucleotidyl transferase was omitted from the enzymatic reaction. The samples were analyzed under a fluorescence microscope (Diaphot 300; Nikon). Four sections per animal were analyzed, and cardiomyocyte nuclei were determined by random counting of 10 fields per section. The number of apoptotic nuclei was calculated per 10,000 cardiomyocytes.
Immunohistochemistry.
Myocardial 3-nitrotyrosine (NY) and 4-hydroxy-2-nonenal (HNE) were assessed, as we have described (27). Briefly, LV tissue sections (4 μm) were blocked with 10% goat serum in phosphate-buffered saline, incubated with rabbit anti-3-nitrotyrosine polyclonal antibody or mouse anti-4-HNE monoclonal antibody, and then incubated with goat biotin-conjugated anti-rabbit IgG or goat biotin-conjugated anti-mouse IgG (Vector Laboratory, Burlingame, CA). The sections were incubated with avidin and biotinylated horseradish peroxidase macromolecular complex (Vector Laboratory) and stained with 3-amino-9-ethylcarbazole (Vector Laboratory) and hematoxylin (Vector Laboratory). For negative control, the primary antibody was omitted instead of normal rabbit IgG or normal mouse IgG. The samples were examined under a light microscopy (BX 40; Olympus). Ten color images of NY or HNE staining were randomly selected from four sections of the heart and photographed at a magnification of ×40. The area and intensity of staining were scored in a blinded manner for quantification as follows: 0, no visible staining; 1, faint staining; 2, moderate staining; and 3, strong staining.
Oxidative post-translational modifications of SERCA.
Oxidative modifications of SERCA were assessed by immonohistochemical staining using site-specific antibodies that detect SERCA sulfonic acid at cysteine 674 or nitrotyrosine at tyrosine 294/295, as we have described in mouse myocardium (18, 36, 38).
SERCA mRNA and protein.
Frozen hearts were ground under liquid nitrogen, and total RNA was extracted with a mirVana miRNA Isolation Kit (Applied Biosystems). Total RNA was treated with DNAse before cDNA synthesis with the High Capacity RNA-to-cDNA Kit (Applied Biosystems). Quantitative PCR was performed with TaqMan Universal PCR Master Mix and TaqMan primers (Applied Biosystems) specific for mouse SERCA2a (Mm01201431_m1) and GAPDH (4352339E) using the Applied Biosystems Step One Plus Real Time PCR System. Data is normalized to GAPDH using the equation 2exp-(CT target gene-CTGAPDH) and expressed as arbitrary units.
Immunoblotting for SERCA protein, hearts were homogenized in lysis buffer (1% Triton X-100, 0.5% Nonidet P-40, 10 mmol/l Tris, 1 mmol/l EDTA, 1 mmol/l EGTA, 150 mmol/l NaCl, 0.4 mmol/l PMSF, 0.2 mmol/l sodium orthovanadate, and 1 g/l leupeptin). Protein concentration was determined using Bradford assay (Bio-Rad). Equal amounts of total protein were separated by SDS-PAGE on 10% gels and transferred to nitrocellulose membrane. The following primary antibodies were used: mouse monoclonal anti-SERCA2 (Affinity Bioreagents) and rabbit polyclonal anti-GAPDH antibody (Abcam). Protein-primary antibody complex was detected by using infrared-dye conjugated goat polyclonal antibody IRDye 680 or IRDye 800 (LICOR Biosciences) and scanned with LI-COR Odyssey Infrared Imaging System.
SERCA activity.
Maximal calcium-stimulated SERCA activity was measured in myocardium as we have described (18). Briefly, LV myocardium was homogenized on ice by sonication in Tris·sucrose homogenization buffer [8% (wt/vol) sucrose in (in mM) 3 Tris·HCl, pH 7.0, 1 PMSF]. The homogenate was centrifuged for 5 min at 4,000 rpm, and the protein concentration of the supernatant was determined by Bradford assay. Samples were pretreated with and without 10 μM of the SERCA inhibitor, thapsigargin. Calcium uptake was initiated by the addition of sample to assay buffer of (in mM) 100 KCl, 5 NaN3, 6 MgCl2, 0.15 EGTA, 0.12 CaCl2, 30 Tris·HCl (pH 7.0), 10 oxalate, and 2.5 ATP containing 1 μCi 45CaCl2 (New England Nuclear, Boston, MA) in a 37°C water bath. Aliquots of each sample taken at 30, 60, and 90 s were vacuum filtered on glass filters (Whatman GF/C; Fisher Scientific, Pittsburgh, PA), washed three times with wash buffer of (in mM) 30 imidazole, 250 sucrose, and 0.5 EGTA and counted with a scintillation counter. SERCA activity is expressed as the rate of thapsigargin-sensitive 45Ca2+ uptake.
Statistical analysis.
Results are presented as means ± SE. The statistical significance of differences among groups or between two means was determined using analysis of variance and the Bonferroni correction for multiple comparisons. Survival was calculated by Kaplan-Meier analysis. P < 0.05 was considered statistically significant.
RESULTS
Myocyte pCAT alleviates myocardial oxidative stress.
Myocardial oxidative stress was assessed by measuring oxidant-mediated HNE and NY protein modifications. In WT mice, AAC caused marked increases in HNE and NY staining, both of which were labeled diffusely over myocytes (Fig. 1). In pCAT mice, the levels of HNE and NY were decreased to levels similar those in sham-operated mice, indicating that increased oxidative stress in mice with AAC is attributable at least in part to H2O2 and is effectively mitigated by pCAT.
Fig. 1.

Immunohistochemical staining for 3-nitrotyrosine (NY) and 4-hydroxy-2-nonenal (HNE) in left ventricular (LV) myocardium of wild-type (WT) or pCAT-over-expressing (CAT) mice subjected to ascending aortic constriction (AAC). NY or HNE staining is shown in red. Nuclei are shown in blue. Bar = 25 μm. The graphs show the relative expression of myocardial NY or HNE in the 4 groups. Values are means ± SE; n = 7–9. *P < 0.001 vs. WT-sham or CAT-sham; †P < 0.005 vs. WT-AAC.
Myocyte pCAT attenuates myocardial hypertrophy.
In WT mice, LV wall thickness measured by echocardiography increased 1 wk after AAC, increased further at 3 wk, and remained constant to slightly decreased over weeks 4 through 12 (Fig. 2). In pCAT mice, increased LV wall thickness with AAC was decreased by ∼32%. Likewise, heart weight (ratio of heart to body weight) in WT mice was increased 12 wk after AAC (vs. sham-operated WT mice), and the increase was attenuated by ∼28% in pCAT mice (Table 1). Myocyte cross-sectional area measured histologically was increased in WT mice with AAC, and the increase was partially (60%) attenuated in pCAT mice (Fig. 3). Myocardial fibrosis, assessed by Masson's trichrome staining, was increased in WT mice with AAC and was markedly decreased in pCAT mice (Fig. 4).
Fig. 2.

A: representative M-mode echocardiograms in WT and CAT mice, 12 wk after AAC or sham surgery. B: changes in LV anterior wall thickness (AWTd), posterior wall thickness (PWTd), total wall thickness (AWTd + PWTd), end-diastolic dimension (EDD) and end-systolic dimension (ESD), and LV fractional shortening (FS) at baseline (BL) before surgery and at 1, 2, 3, 4, 8, and 12 wk (W) after surgery. Values are means ± SE; n = 9–20. *P < 0.02 vs. WT-sham or CAT-sham.; †P < 0.05 vs. WT-AAC.
Table 1.
Body and organ weights
| WT-Sham | Catalase-Sham | WT-AAC | Catalase-AAC | |
|---|---|---|---|---|
| Body weight, g | 29.8 ± 0.5 | 30.1 ± 1.1 | 27.4 ± 1.4 | 27.2 ± 1.5 |
| Heart weight, mg | 139.8 ± 2.9 | 139.5 ± 3.7 | 237.4 ± 10.5* | 191.9 ± 12.0*† |
| Heart weight/body weight, mg/g | 4.43 ± 0.10 | 4.51 ± 0.11 | 8.93 ± 0.74* | 6.98 ± 0.48*† |
| Lung weight, mg | 174.5 ± 9.3 | 168.1 ± 4.3 | 261.5 ± 35.6* | 186.3 ± 18.4 |
| Lung weight/body weight, mg/g | 5.51 ± 0.27 | 5.44 ± 0.14 | 10.20 ± 1.87* | 5.99 ± 0.24† |
| Lung weight, wet/dry | 4.67 ± 0.09 | 4.84 ± 0.10 | 5.14 ± 0.21* | 4.68 ± 0.07† |
| Liver weight, g | 1.38 ± 0.04 | 1.35 ± 0.53 | 1.27 ± 0.61 | 1.27 ± 0.28 |
| Liver weight/body weight, g/g | 0.046 ± 0.002 | 0.044 ± 0.001 | 0.046 ± 0.001 | 0.047 ± 0.001 |
| Pleural effusion | 0/10 | 0/11 | 7/9* | 1/15† |
Values are means ± SE; n = 10–15.
P < 0.05 vs. wild-type (WT)-sham;
P < 0.05 vs. WT-ascending aortic constriction (AAC).
Fig. 3.

Myocyte size in WT and CAT mice 12 wk after surgery. Top: representative photomicrographs of LV sections stained by hematoxyline and eosin. Bar (WT-sham panel) = 25 μm. Bottom: quantification of myocyte cross-sectional area measured using NIH ImageJ. Values are means ± SE; n = 6–8. *P < 0.005 vs. WT-sham or CAT-sham; #P < 0.05 vs. WT-sham or CAT-sham; †P < 0.02 vs. WT-AAC.
Fig. 4.

Representative photomicrographs of LV myocardium with Masson trichrome staining for fibrosis 12 wk after surgery. Myocardium stains red, and collagen stains blue. Bar = 100 μm. Bottom: quantification of cardiac fibrosis measured using NIH ImageJ. Values are means ± SE; n = 6–8. *P < 0.001 vs. WT-sham or CAT-sham; #P < 0.05 vs. WT-sham or CAT-sham; †P < 0.001 vs. WT-AAC.
Myocyte pCAT prevents progression to LV failure.
In WT mice, ACC caused progressive LV dilation and systolic failure as assessed by echocardiography. LV EDD and LV ESD increased progressively beginning 1 to 2 wk after AAC, in association with a decrease in LV FS (Fig. 2). Myocyte-specific overexpression of pCAT attenuated both LV dilation and the decrease in FS. Myocyte apoptosis, a major mechanism responsible for LV failure with AAC (10), was assessed by measurement of TUNEL staining. The frequency of TUNEL-positive myocytes increased approximately eightfold in WT mice after AAC, and the increase was markedly decreased by pCAT (Fig. 5).
Fig. 5.

A: representative photomicrographs of LV showing terminafl deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) staining for apoptotic myocytes. Apoptotic nuclei (arrows) are shown by green fluorescence in a, e, i, and m. Nuclei (arrowheads) stained by Hoechst 33258 are shown blue in b, f, j, and n. Cardiomyocytes identified by α-sarcomeric actin staining are shown red in c, g, k, and o. The overlays in d, h, l, and p allow identification of apoptotic nuclei present in myocytes. Bar (A) = 25 μm. B: mean changes in the number of apoptotic myocytes in LV myocardium 12 wk after surgery. Values are means ± SE; n = 5–7. *P < 0.001 vs. WT-sham or CAT-sham; #P < 0.05 vs. WT-sham or CAT-sham; †P < 0.002 vs. WT-AAC.
In WT mice, AAC caused functional heart failure as reflected by an ∼60% decrease in maximal exercise capacity, which was partially corrected by pCAT (Fig. 6). Lung weight (wet/dry) was increased by AAC in WT mice, indicative of lung congestion, and the increase was attenuated in pCAT mice (Table 1). Likewise, pleural effusions were present in 90% of WT mice surviving 12 wk after AAC, but were present in only 6% of pCAT mice, and absent in sham-operated WT or pCAT mice. Survival rate at 12 wk was 100% in sham-operated mice, 45% in WT with AAC, and was increased to 75% in pCAT mice with AAC (Fig. 6).
Fig. 6.
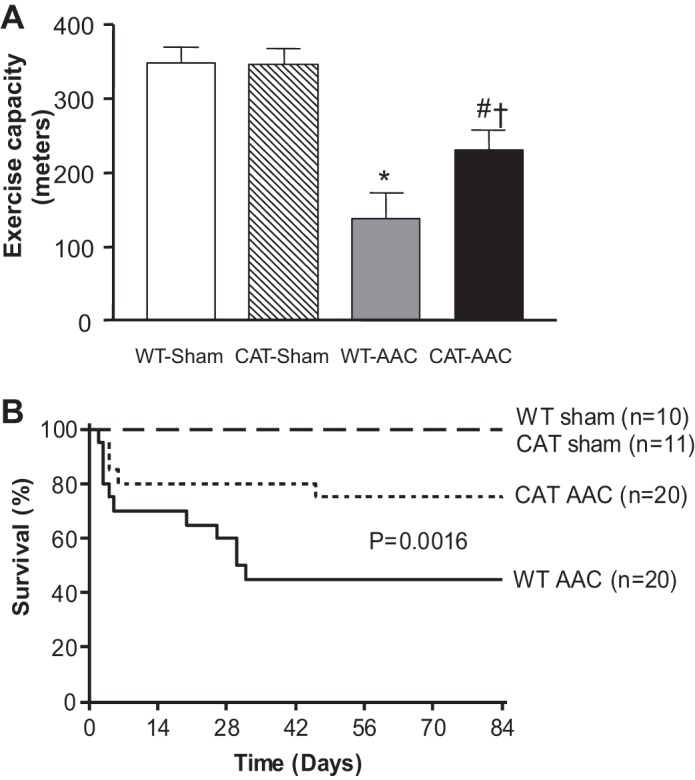
A: maximal exercise capacity 12 wk after surgery. Values are means ± SE; n = 7–11. *P < 0.005 vs. WT-sham or CAT-sham; #P < 0.05 vs. WT-sham or CAT-sham; †P < 0.05 vs. WT-AAC. B: Kaplan-Meier survival analysis through 12 wk when mice were euthanized.
Myocyte pCAT prevents oxidative modification of SERCA.
Oxidative modifications of SERCA were assessed immunohistologically using site-specific antibodies for oxidatively modified SERCA with sulfonic acid at cysteine 674 (SERCA-C674-SO3H) (38) or nitrotyrosine 294/295 (SERCA-N294/295-Y) (36). In sham-operated mice there were low levels of both SERCA-C674-SO3H and SERCA-N294/295-Y, and both levels were markedly increased in WT mice with AAC (Fig. 7). In pCAT mice with AAC, both oxidative modifications of SERCA were decreased to the low levels present in sham-operated mice.
Fig. 7.

Representative photomicrographs of LV myocardium subjected to immunohistochemical staining with site-specific antibodies that recognize sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) that is nitrated at tyrosine 294/295 (SERCA-NY 294,295; A) or sulfonated on C674 (SERCA C674-SO3; B). Bar (WT-sham) = 25 μm.
Myocyte pCAT preserves SERCA activity.
We have shown that oxidative modifications of SERCA are associated with decreases in Ca2+ uptake activity (18, 28, 38). Therefore, SERCA activity was assessed by measuring maximal calcium-stimulated 45Ca2+ uptake. In WT mice, AAC caused a ∼46% decrease in SERCA activity (vs. sham-operated mice), which was restored, at least partially, in pCAT mice (Fig. 8A).
Fig. 8.
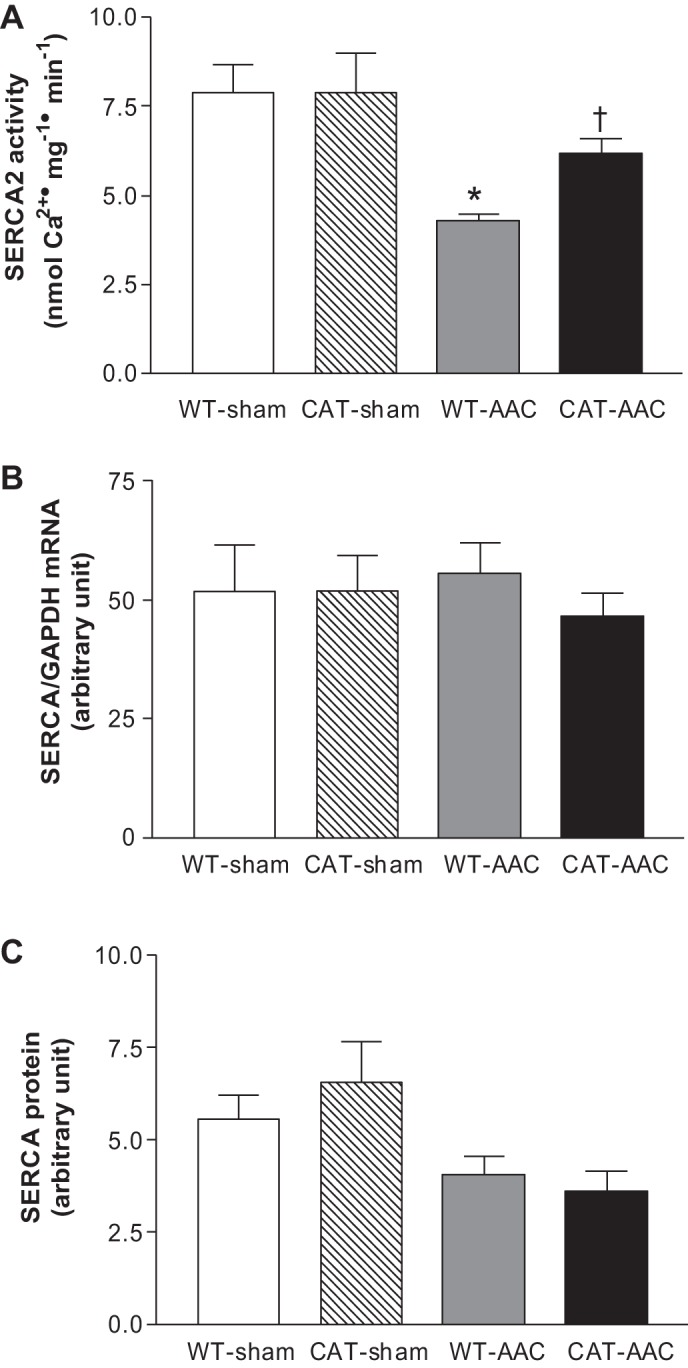
Maximal calcium-stimulated SERCA activity (A), SERCA mRNA (B), and SERCA protein expression (C) measured in LV myocardium 12 wk after surgery. Values are means ± SE; n = 3 for SERCA activity and n = 4 for SERCA mRNA or protein expression. *P < 0.01 vs. WT-sham or CAT-sham; †P < 0.01 vs. WT-AAC.
SERCA mRNA levels were not affected by AAC or pCAT (Fig. 8B). SERCA protein levels were decreased by AAC but were not affected further by pCAT (Fig. 8C), suggesting that the increase in SERCA activity with pCAT is mediated at the post-translational level.
DISCUSSION
There are two major new findings in this study. First, we show for the first time that cardiac myocyte-restricted expression of pCAT effectively ameliorates the structural and functional consequences of chronic hemodynamic overload. This finding suggests that sarcoplasmic ROS in the cardiac myocyte plays a central role in mediating the effects of hemodynamic overload. Second, we show for the first time that chronic hemodynamic overload causes oxidative modifications of SERCA that are associated with decreased activity and that both SERCA oxidation and function are improved by pCAT in the absence of changes in transcript or protein expression. This finding provides a new mechanistic basis for impaired calcium regulation and myocyte function in chronic hemodynamic overload, and further identifies SERCA as an important target of ROS that can be mitigated by sarcoplasmic pCAT.
pCAT effectively inhibits hypertrophy and progression to failure.
In WT mice AAC initially led to LV hypertrophy followed by progressive chamber dilation and contractile failure, associated at the cellular level with myocyte hypertrophy and apoptosis. LV hypertrophy and failure were inhibited in pCAT-expressing mice, as were myocyte hypertrophy and apoptosis. The functional importance of these effects of pCAT is supported by the concomitant findings of improved exercise function, decreased lung congestion, and improved survival. Thus pCAT effectively ameliorates the effects of hemodynamic overload on myocardial structure and function.
The effects of pCAT that we observed are qualitatively and quantitatively similar to those observed in this model in transgenic mice with total body overexpression of mCAT (6). The localization of catalase in the pCAT mouse used in our study was determined by electron microscopy, which showed that the pCAT is expressed primarily in peroxisomes, sarcoplasm, and nucleus but is not expressed in mitochondria (39). Conversely, the mCAT mouse was shown to express catalase only in mitochondria (7). The comparable effectiveness of mCAT and pCAT in pressure overload suggests that both catalase locations have access to ROS that ultimately reaches critical target molecules located in the sarcoplasm. These experiments do not identify the source of the ROS, which might originate in mitochondria (7) and/or from one or more sarcoplasmic sources including uncoupled endothelial nitric oxide synthase (33), NOX2 (13), or xanthine oxidase (37). However, because pCAT does not enter mitochondria our data indicate that scavenging of ROS in the sarcoplasm is sufficient to ameliorate important aspects of the phenotype including hypertrophy, apoptosis, and impaired SERCA activity.
This is the first demonstration of the effects of pCAT in AAC or any other model of hemodynamic overload. The transgenic pCAT mouse used in these experiments was one of several lines (line 742) developed by Kang et al. (16) and shown to oppose the cardiac effects of doxorubicin (16) and ischemia-reperfusion (20). We found that the pCAT mouse ameliorates the pathologic phenotype in Gαq-overexpressing mice (5, 27). Although we did not assess the antioxidant effectiveness of pCAT in this study, in the Gaq mouse pCAT corrected the GSH-to-GSSG ratio to normal (27). Our observations in the Gαq mouse differ from a report by Dai et al. (7) who found no benefit with a different pCAT mouse. These discrepant results may reflect the use in their experiments of a different line of pCAT mice (line 776) and/or the mixed background of their mice as opposed to the pure FVB background in our study. We also found that pCAT mice have less LV hypertrophy and diastolic dysfunction with age (28).
pCAT protects SERCA function.
A major finding of this study is that decreased SERCA activity after AAC was partially prevented by pCAT. Myocardial SERCA activity is decreased in pressure overload leading to sarcoplasmic reticulum calcium depletion, abnormal calcium transients, and impaired systolic and diastolic function at the myocyte level (15, 26). These abnormalities are ameliorated by transgenic overexpression of WT SERCA (14, 25) and worsened in SERCA+/− mice (31). Likewise, viral expression of SERCA improves LV function in rats with hemodynamic overload (22), aging (30), and other models of heart failure (24), and has led to the investigational use of virally mediated SERCA expression in patients with heart failure (40). Decreased SERCA activity in pressure overload has generally been attributed to decreased protein expression (26), and consistent with prior observations, the expression of SERCA protein was decreased with AAC in our study. However, because neither SERCA protein nor mRNA expression was affected by pCAT, the pCAT-induced increase in activity cannot be attributed to increased SERCA levels. In contrast with most other studies in mice with aortic constriction (15), in our study the decrease in SERCA protein was not associated with a decrease in mRNA, possibly reflecting increased protein degradation.
SERCA activity can also be regulated by OPTM. Low concentrations of ROS activate SERCA via reversible glutathiolation of cysteine 674 (2, 19), and this activation is inhibited by higher concentrations of ROS that irreversibly oxidize this cysteine (18, 28, 38). Using site-directed antibodies to identify sulfonic acid at SERCA cysteine 674 and nitrotyrosine at tyrosine-294/5, we found that AAC increases both oxidative modifications, and that both are prevented by pCAT. The observation that pCAT corrects SERCA OPTM and restores SERCA activity in mice with AAC, without increasing SERCA expression (i.e., WT-AAC vs. CAT-AAC), strongly supports the thesis that pCAT increases SERCA activity by decreasing inhibitory oxidative modifications. The ability of pCAT to decrease SERCA oxidation is consistent with the ability of H2O2 to diffuse across membranes, and in this case, into peroxisomes, which may act as an antioxidant 'sink′ in the sarcoplasm.
In summary, the ability of pCAT to protect the myocardium from the cardinal features of hemodynamic overload supports the utility of protecting sarcoplasmic targets from ROS. In this regard, we show that SERCA is an important target of ROS that contributes to the pathophysiology of hemodynamic overload and is protected by pCAT. Improved SERCA function has potentially important consequences for cardiac excitation/contraction (15). In addition, by affecting endoplasmic reticulum calcium stores, SERCA may play a role in a variety of other biologic processes including endoplasmic reticulum stress, the unfolded protein response, growth signaling, and apoptosis (11). These findings expand our understanding of the mechanisms by which ROS mediate heart failure, suggest that prevention of target protein oxidation may be a useful therapeutic approach, and have implications for the optimal repletion of SERCA in failing myocardium.
GRANTS
This work was supported by NIH Grants HL-061639 (to W. S. Colucci), HL-064750 (to W. S. Colucci), HL-031607 (to R. A. Cohen), PO1 HL-068758 (to R. A. Cohen), and R37-104017 (to R. A. Cohen) and the National Heart, Lung, and Blood Institute-sponsored Boston University Cardiovascular Proteomics Center (Contract No. N01-HV-28178; to R. A. Cohen and W. S. Colucci).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: F.Q., D.R.P., Y.J.K., R.A.C., and W.S.C. conception and design of research; F.Q., D.A.S., R.J.M., A.B., and V.H.T. performed experiments; F.Q., D.A.S., R.J.M., A.B., and V.H.T. analyzed data; F.Q., D.A.S., D.R.P., R.A.C., and W.S.C. interpreted results of experiments; F.Q., A.B., and V.H.T. prepared figures; F.Q. drafted manuscript; F.Q., D.A.S., D.R.P., R.J.M., A.B., V.H.T., Y.J.K., R.A.C., and W.S.C. edited and revised manuscript; F.Q., D.A.S., D.R.P., R.J.M., A.B., V.H.T., Y.J.K., R.A.C., and W.S.C. approved final version of manuscript.
REFERENCES
- 1.Adachi T, Matsui R, Xu S, Kirber M, Lazar HL, Sharov VS, Schoneich C, Cohen RA. Antioxidant improves smooth muscle sarco/endoplasmic reticulum Ca2+-ATPase function and lowers tyrosine nitration in hypercholesterolemia and improves nitric oxide-induced relaxation. Circ Res 90: 1114–1121, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 10: 1200–1207, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Biolo A, Greferath R, Siwik DA, Qin F, Valsky E, Fylaktakidou KC, Pothukanuri S, Duarte CD, Schwarz RP, Lehn JM, Nicolau C, Colucci WS. Enhanced exercise capacity in mice with severe heart failure treated with an allosteric effector of hemoglobin, myo-inositol trispyrophosphate. Proc Natl Acad Sci USA 106: 1926–1929, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 93: 802–805, 2003 [DOI] [PubMed] [Google Scholar]
- 5.D′Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., 2 Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA 94: 8121–8126, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res 93: 79–88, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 108: 837–846, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Date MO, Morita T, Yamashita N, Nishida K, Yamaguchi O, Higuchi Y, Hirotani S, Matsumura Y, Hori M, Tada M, Otsu K. The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model. J Am Coll Cardiol 39: 907–912, 2002 [DOI] [PubMed] [Google Scholar]
- 9.del M, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 100: 2308–2311, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding B, Price RL, Goldsmith EC, Borg TK, Yan X, Douglas PS, Weinberg EO, Bartunek J, Thielen T, Didenko VV, Lorell BH. Left ventricular hypertrophy in ascending aortic stenosis mice: anoikis and the progression to early failure. Circulation 101: 2854–2862, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: programmed to conserve. J Mol Cell Cardiol 55: 85–91, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisner D, Caldwell J, Trafford A. Sarcoplasmic reticulum Ca-ATPase and heart failure 20 years later. Circ Res 113: 958–961, 2013 [DOI] [PubMed] [Google Scholar]
- 13.Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, Shah AM. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J Am Coll Cardiol 47: 817–826, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Ito K, Yan X, Feng X, Manning WJ, Dillmann WH, Lorell BH. Transgenic expression of sarcoplasmic reticulum Ca2+ ATPase modifies the transition from hypertrophy to early heart failure. Circ Res 89: 422–429, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Ito K, Yan X, Tajima M, Su Z, Barry WH, Lorell BH. Contractile reserve and intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing hearts. Circ Res 87: 588–595, 2000 [DOI] [PubMed] [Google Scholar]
- 16.Kang YJ, Chen Y, Epstein PN. Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J Biol Chem 271: 12610–12616, 1996 [DOI] [PubMed] [Google Scholar]
- 17.Kuster GM, Kotlyar E, Rude MK, Siwik DA, Liao R, Colucci WS, Sam F. Mineralocorticoid receptor inhibition ameliorates the transition to myocardial failure and decreases oxidative stress and inflammation in mice with chronic pressure overload. Circulation 111: 420–427, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ Res 107: 228–232, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lancel S, Zhang J, Evangelista A, Trucillo MP, Tong X, Siwik DA, Cohen RA, Colucci WS. Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ Res 104: 720–723, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li G, Chen Y, Saari JT, Kang YJ. Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 273: H1090–H1095, 1997 [DOI] [PubMed] [Google Scholar]
- 21.Minhas KM, Saraiva RM, Schuleri KH, Lehrke S, Zheng M, Saliaris AP, Berry CE, Barouch LA, Vandegaer KM, Li D, Hare JM. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res 98: 271–279, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci USA 97: 793–798, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moens AL, Leyton-Mange JS, Niu X, Yang R, Cingolani O, Arkenbout EK, Champion HC, Bedja D, Gabrielson KL, Chen J, Xia Y, Hale AB, Channon KM, Halushka MK, Barker N, Wuyts FL, Kaminski PM, Wolin MS, Kass DA, Barouch LA. Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure-overload in mice lacking the beta3-adrenoreceptor. J Mol Cell Cardiol 47: 576–585, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monte FD, Hajjar RJ. Targeting calcium cycling proteins in heart failure through gene transfer. J Physiol 546: 49–61, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller OJ, Lange M, Rattunde H, Lorenzen HP, Muller M, Frey N, Bittner C, Simonides W, Katus HA, Franz WM. Transgenic rat hearts overexpressing SERCA2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res 59: 380–389, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Periasamy M, Bhupathy P, Babu GJ. Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc Res 77: 265–273, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Qin F, Lennon-Edwards S, Lancel S, Biolo A, Siwik DA, Pimentel DR, Dorn GW, Kang YJ, Colucci WS. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and -independent phases of myocardial remodeling and prevents the progression to overt heart failure in Gαq-overexpressing transgenic mice. Circ Heart Fail 3: 306–313, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin F, Siwik DA, Lancel S, Zhang J, Kuster GM, Luptak I, Wang L, Tong X, Kang YJ, Cohen RA, Colucci WS. Hydrogen peroxide-mediated SERCA cysteine 674 oxidation contributes to impaired cardiac myocyte relaxation in senescent mouse heart. J Am Heart Assoc 2: e000184, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol 34: 379–388, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Schmidt U, del MF, Miyamoto MI, Matsui T, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum Ca2+-ATPase. Circulation 101: 790–796, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Schultz JJ, Glascock BJ, Witt SA, Nieman ML, Nattamai KJ, Liu LH, Lorenz JN, Shull GE, Kimball TR, Periasamy M. Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am J Physiol Heart Circ Physiol 286: H1146–H1153, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 93: 903–907, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest 115: 1221–1231, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 49: 241–248, 2007 [DOI] [PubMed] [Google Scholar]
- 35.van Empel VP, Bertrand AT, van Oort RJ, van der NR, Engelen M, van Rijen HV, Doevendans PA, Crijns HJ, Ackerman SL, Sluiter W, De Windt LJ. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol 48: 824–832, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Xu S, Ying J, Jiang B, Guo W, Adachi T, Sharov V, Lazar H, Menzoian J, Knyushko TV, Bigelow D, Schoneich C, Cohen RA. Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol 290: H2220–H2227, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Xu X, Hu X, Lu Z, Zhang P, Zhao L, Wessale JL, Bache RJ, Chen Y. Xanthine oxidase inhibition with febuxostat attenuates systolic overload-induced left ventricular hypertrophy and dysfunction in mice. J Card Fail 14: 746–753, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ying J, Sharov V, Xu S, Jiang B, Gerrity R, Schoneich C, Cohen RA. Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2+) ATPase in diabetic pig aorta. Free Radic Biol Med 45: 756–762, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Kang YJ. Cellular and subcellular localization of catalase in the heart of transgenic mice. J Histochem Cytochem 48: 585–594, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Zsebo KM, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ. Long term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res 114: 101–108, 2014 [DOI] [PubMed] [Google Scholar]