

Abstract
Renal blood flow (RBF) responses to arginine vasopressin (AVP) were tested in anesthetized wild-type (WT) and CD38−/− mice that lack the major calcium-mobilizing second messenger cyclic ADP ribose. AVP (3–25 ng) injected intravenously produced dose-dependent decreases in RBF, reaching a maximum of 25 ± 2% below basal RBF in WT and 27 ± 2% in CD38−/− mice with 25 ng of AVP. Renal vascular resistance (RVR) increased 75 ± 6% and 78 ± 6% in WT and CD38−/− mice. Inhibition of nitric oxide (NO) synthase with nitro-l-arginine methyl ester (l-NAME) increased the maximum RVR response to AVP to 308 ± 76% in WT and 388 ± 81% in CD38−/− (P < 0.001 for both). Cyclooxygenase inhibition with indomethacin increased the maximum RVR response to 125 ± 15% in WT and 120 ± 14% in CD38−/− mice (P < 0.001, <0.05). Superoxide suppression with tempol inhibited the maximum RVR response to AVP by 38% in both strains (P < 0.005) but was ineffective when administered after l-NAME. The rate of RBF recovery (relaxation) after AVP was slowed by l-NAME and indomethacin (P < 0.001, <0.005) but was unchanged by tempol. All vascular responses to AVP were abolished by an AVP V1a receptor antagonist. A V2 receptor agonist or antagonist had no effect on AVP-induced renal vasoconstriction. Taken together, the results indicate that renal vasoconstriction by AVP in the mouse is strongly buffered by vasodilatory actions of NO and prostanoids. The vasoconstriction depends on V1a receptor activation without involvement of CD38 or concomitant vasodilatation by V2 receptors. The role of superoxide is to enhance the contractile response to AVP, most likely by reducing the availability of NO rather than directly stimulating intracellular contraction signaling pathways.
Keywords: arginine vasopressin, indomethacin, nitro-l-arginine methyl ester, tempol, renal blood flow, renal vascular resistance, prostaglandin E2, prostaglandin I2, superoxide dismutase, reactive oxygen species, V1a receptor
arginine vasopressin (AVP) exerts diverse actions on the kidney and the cardiovascular system. In the kidney, AVP promotes water reabsorption by stimulating V2 receptors that regulate aquaporin 2 in the distal tubule and collecting duct (50) and increase sodium reabsorption in distal segments beginning with the ascending limb of Henle's loop (72, 78). Cardiovascular actions of AVP are primarily mediated by V1a receptors that induce vasoconstriction through activation of Gq11/12-coupled receptors (51). In some species, such as dog and rabbit, AVP also stimulates V2 receptors in the vasculature to produce vasodilation (2, 55, 67, 82). V2 receptors may inhibit V1a-mediated constriction of rat descending vasa recta (86). The receptors mediating the effects of AVP on the renal vasculature of the mouse are not known.
Mice with V1a receptors genetically deleted are characterized by attenuated vasoconstrictor responses to AVP, hypotension, reduced blood volume, decreased sympathetic nerve activity, and reduced activity of the renin-angiotensin-aldosterone system (4, 51). These mice also show decreased susceptibility to salt-induced hypertension (69). Intravenous or intrarenal administration of a V1 receptor agonist produces hypertension in previously normotensive rats (21, 22, 79, 80). In models of genetic hypertension, such as the spontaneously hypertensive rat (SHR) and the SHR-stroke-prone rat (SHR-SP), AVP causes exaggerated renal vasoconstriction compared with normotensive controls (11, 30, 32) and contributes to the elevated arterial pressure (17, 64, 77). AVP has been implicated in the pathophysiology of hypertension in humans and appears to contribute more to essential hypertension in African-Americans than Caucasian subjects (7).
In common with other vasoconstrictor G protein-coupled receptors (GPCRs), the response to AVP V1a receptor activation in vascular smooth muscle cells (VSMC) usually involves enhanced Ca2+ entry across the plasma membrane in combination with Ca2+ mobilization from internal sarcoplasmic reticulum stores (25, 26, 32, 42, 81). In addition to these mechanisms, many GPCRs that initiate renal vasoconstriction, e.g., AT1 receptors for angiotensin II and ETA endothelin-1 receptors, also stimulate the ADP ribosyl cyclase CD38 to activate Ca2+ mobilization via ryanodine receptors (RyR) in VSMC (28, 29). At present, it is not known whether renal vascular responsiveness to AVP also involves the CD38/RyR/Ca2+ signaling pathway.
Vasodilators including nitric oxide (NO) and cyclooxygenase (COX)-derived prostanoids usually blunt the magnitude of AVP-induced vasoconstriction (8, 36, 41, 57, 70), and the arterial baroreflex strongly counteracts AVP-mediated increases in arterial pressure (AP) (37). The renal circulation appears to be particularly well buffered against the vasoconstrictor actions of AVP (36), with participation of either V1a or both V1a and V2 receptors, depending on species (2, 55, 67, 82, 86).
To our knowledge, no study has evaluated mechanisms mediating AVP-induced renal vasoconstriction in the mouse. The purpose of our study was to test whether the renal vascular actions of AVP are mediated by V1 or a combination of V1 and V2 receptors. A second aim was to determine the extent to which the ADP ribosyl cyclase CD38 contributes to AVP-induced renal vasoconstriction by testing responses in wild-type (WT) and CD38-null mice. We also tested the effects of NO synthase (NOS) and COX inhibition to determine the extent to which NO or vasodilator prostanoids buffer the magnitude of acute renal vasoconstriction produced by AVP. Other experiments assessed the effects of superoxide (O2·−) dismutation with tempol to determine the contribution of reactive oxygen species to the renal vascular actions of AVP.
MATERIALS AND METHODS
CD38 knockout mice and C57BL6J WT mice were bred in our animal facility from original stock provided by Dr. F. Lund (84). Animals were housed and studied in accordance with the NIH guidelines for care and use of laboratory animals, and all experimental protocols were approved by the Animal Care and Use Committee at the University of North Carolina at Chapel Hill. Tap water and chow containing 0.4% sodium were provided ad libitum. Animals were not fasted before study.
Animal preparation.
Animals were anesthetized with pentobarbital sodium (80 mg/kg ip) and placed on a thermostatically controlled operating table to maintained body temperature at 37°C (65). A tracheotomy tube (PE90) was installed to maintain a clear airway and the urinary bladder cannulated at the dome with PE50 tubing for urine drainage. Maintenance infusions of 0.9% NaCl at 0.3 μl/min per g body wt and a 2% solution of BSA in 0.9% NaCl at 3 μl/min were provided through indwelling cannulas in the right jugular vein. Anesthesia was sustained with intermittent supplemental intravenous doses of pentobarbital. The left carotid artery was cannulated with a narrowed PE50 cannula for measurement of AP with a pressure transducer (Statham Gould P23Db) connected to a carrier amplifier (HP 8805B). The right kidney was exposed in the retroperitoneal space through a flank incision and the renal artery separated from the renal vein and surrounding adventitia. As part of the renal artery dissection, the renal nerves were severed to isolate the renal circulation from sympathetic nerve activity. A transit time blood flow transducer (Transonic MA0.5PSB) was positioned around the renal artery and connected to a flow meter (Transonic TS420). AP and RBF signals were digitized at 120 Hz with an A/D converter (DT9816) for continuous display on a PC-based data acquisition system (DtEz software). Data were averaged into 1-s bins and stored on a disk for offline analysis.
Precise 10-μl boluses of AVP at doses of 3, 5, 10, and 25 ng (equivalent to 3, 5, 10, and 25 pmol, or ∼120, 200, 400, and 1,000 pmol/kg) were injected into the jugular vein with a high-performance liquid chromatography injection valve positioned in line with the 2% BSA infusion. For each injection, the infusion rate was increased from 3 μl/min to 60 μl/min simultaneously with activation of the injection valve and then returned to 3 μl/min 30 s later. Each AVP injection was complete 20 s after activation of the valve. The time between individual injections was based on the return of RBF and AP to preinjection levels but was at least 10 min for all injections. At the end of each experiment, an animal was euthanized with an overdose of anesthetic, and the right kidney was removed, decapsulated, and weighed.
Experimental protocols.
After completion of surgery, 30–45 min were allowed for stabilization before AVP injections commenced. All doses were tested in duplicate in a randomized sequence during a control period followed by an experimental period that evaluated the effects of NO depletion with l-NAME (25 mg/kg iv), COX inhibition with indomethacin (170 μg/kg per min), or O2·− suppression with tempol (4 mg/kg per min). These agents were infused for 20–30 min before AVP injections were recommenced. In a subgroup of nine mice receiving l-NAME, a third period of AVP injections was included to test the effects of tempol after inhibition of NOS with l-NAME. Acute inhibition of normally coupled NOS with l-NAME effectively reduces renal production of NO and urinary nitrite/nitrate excretion and causes renal vasoconstriction in healthy animals (60, 61, 92). In vitro studies have shown that NOS inhibition reduces endothelium-dependent NO production by isolated renal vessels (71, 99, 101).
The contribution of V1a receptor activation to renal vascular responses to AVP was tested in a separate group of WT mice in which the effects of AVP were determined before and during an intravenous infusion of the selective V1a receptor antagonist Manning compound [β-Mercapto-β,β-cyclopentamethylenepropionyl1, O-me-Tyr2, Arg8]-Vasopressin (MC, Sigma) (62). In accordance with published reports, MC was infused at 100 ng/min iv to achieve effective blockade of peripheral V1a receptors in mice (1). In some animals, antagonism of V2 receptors [Adamantaneacetyl1, O-Et-D-Tyr2, Val4, Aminobutyryl6, Arg8,9]-Vasopressin (62) was tested in combination with V1a receptor blockade. In other animals, V2 receptor activation was tested in a group of five WT mice in which RBF and AP were monitored during intravenous bolus injections (50 and 100 ng) of the selective V2 receptor agonist dDAVP (desmopressin) (91).
Analysis of results.
A 300-s sampling interval was allocated to each AVP injection, beginning with a 60-s baseline period immediately before the onset of each vasoconstrictor response. Values for mean arterial pressure (MAP), RBF, and RVR were converted to a percentage change from the mean values obtained for each parameter during the 60-s baseline period. The magnitude of vasoconstrictor responses to AVP were evaluated as the maximum percent change from baseline and from the integrated area of each response curve over a 120-s period beginning at the onset of the response. Dynamic characteristics of responses to AVP were estimated from the downslope (vasoconstrictor phase) and upslope (relaxation phase) of the RBF response. These rates were obtained from the first derivative of the RBF data trace over linear segments of the contractile and relaxation phases of the response. A linear portion in the downslopes consistently occurred between 5 and 8 s after the beginning of each response, and a straight segment in the upslope occurred between 10 and 20 s after the maximum reduction in RBF. The response slopes were expressed as %RBFchange/s compared with the baseline RBF and plotted against the corresponding mean maximum percent reduction in RBF for each dose.
The Prism statistical and graphing software program was used for data analyses and presentation (v.6.03 GraphPad). Overall responses are reported as means ± SE for each dose. Comparisons within groups (control vs. experimental periods) and between groups (WT vs. CD38−/−) were performed with paired and unpaired t-tests or two-way analysis of variance. Dose-response curves for maximum percent changes in AP, RBF, RVR, and rates of contraction or relaxation were analyzed with nonlinear regression using best-fit curves, which were either straight lines or one-phase exponentials, depending on the data.
RESULTS
Baseline values during control conditions for 30 WT and 30 CD38-deficient mice are presented in Table 1. MAP, RBF, and RVR were similar in the two groups, as were body weight and left kidney weight. Hematocrit was stable throughout each experiment.
Table 1.
Basal values for WT and CD38−/− mice
| WT | CD38−/− | |
|---|---|---|
| Arterial blood pressure, mmHg | 109 ± 1 | 107 ± 2 |
| Heart rate, beats/min | 465 ± 11 | 441 ± 14 |
| Renal blood flow, ml·min−1·g kidney wt−1 | 8.42 ± 0.47 | 9.23 ± 0.57 |
| Renal vascular resistance, mmHg·ml−1·min−1·g kidney wt | 14.7 ± 1.3 | 12.7 ± 0.9 |
| Body weight, g | 26.4 ± 0.4 | 25.1 ± 0.6 |
| Right kidney weight, g | 0.192 ± 0.003 | 0.189 ± 0.004 |
| HCT-1, % | 43.5 ± 1.0 | 41.6 ± 1.0 |
| HCT-2, % | 44.0 ± 1.3 | 43.9 ± 1.3 |
| Number of animals | 30 | 30 |
Values are means ± SE. There were no significant differences between strains. HCT-1, start hematocrit; HCT-2, end hematocrit; WT, wild-type.
Vascular responses to bolus injections of AVP in WT mice are shown in Fig. 1, top. After a 60-s baseline recording, AVP was injected intravenously, and the hemodynamic responses were recorded for the following 240 s. A dose-response relationship was apparent for MAP and RVR at all doses of AVP and for RBF at the two higher doses (10 and 25 ng). All AVP doses resulted in a small increase in RBF after 120 s, which was accompanied by a persistent increase in RVR. This combination made it most likely that the increase in RBF is the result of a relatively prolonged systemic vasopressor action of AVP compared with a more rapid decline in renal vasoconstriction. Figure 1, bottom, shows the maximum responses produced by AVP during control conditions (○). RBF was reduced in a dose-dependent manner to a maximum of 28% below baseline. RVR progressively increased to 75% above control, whereas MAP increased 35%.
Fig. 1.

Arginine vasopressin (AVP)-induced changes in renal blood flow (RBF), renal vascular resistance (RVR), and mean arterial pressure (MAP) in the control period in wild-type (WT) mice (n = 20). Top: mean responses to 4 doses of AVP (3–25 ng) administered at 60 s. For clarity, symbols are shown at 10-s intervals; SE is shown by dashed lines. Bottom: maximum changes recorded during the control period (○) for each dose of AVP. ■, maximum responses to the same doses of AVP following V1a receptor antagonism with Manning compound [β-Mercapto-β,β-cyclopentamethylenepropionyl1, O-me-Tyr2, Arg8]-Vasopressin (n = 6). ▲, responses to V2 receptor activation with dDAVP (desmopressin, 25 and 50 ng, n = 5).
Renal and systemic vascular responses to bolus injections of the V2 receptor agonist are also shown in Fig. 1 (bottom, ▲). The small changes in RBF and RVR (<5% of baseline) were not different from vehicle injections. These data show no sign of a vasodilatory response to dDAVP at the renal or systemic level.
The V1a receptor antagonist MC was administered to WT mice (n = 8) to evaluate the contribution of this receptor to the action of endogenous AVP and to the acute hemodynamic responses to administered AVP. Baseline MAP was not affected by MC, but heart rate was increased (Table 2). RBF rose by 15% (P < 0.05), and RVR fell by 7% (P < 0.05), which is consistent with a small to modest effect of endogenous AVP on the renal vasculature during anesthesia. Importantly, the renal vasoconstriction normally produced by administered AVP was abolished by V1a receptor blockade with MC (Fig. 1, bottom, ■). MC also almost completely eliminated the pressor response to AVP. These results suggest that the renal vascular effects of AVP are mediated exclusively by V1a receptors in the mouse. This conclusion is reinforced by the finding that antagonism of V2 receptors with [Adamantaneacetyl1, O-Et-D-Tyr2, Val4, Aminobutyryl6, Arg8,9]-Vasopressin (dose 2–4 nmol/kg) (62) during continued blockade of V1a receptors had no effect on RBF or RVR (data not shown).
Table 2.
Basal values before (control) and during AVP V1a receptor blockade with MC in WT mice
| Control | V1a Receptor Blockade | |
|---|---|---|
| Arterial blood pressure, mmHg | 113 ± 4 | 115 ± 3 |
| Heart rate, beats/min | 474 ± 37 | 560 ± 53† |
| Renal blood flow, ml·min−1·g kidney wt−1 | 7.93 ± 1.20 | 9.13 ± 1.5* |
| Renal vascular resistance, mmHg·ml−1·min−1·g kidney wt | 19.5 ± 4.8 | 18.2 ± 4.7* |
| Number of animals | 8 | 8 |
Values are means ± SE. Control and Manning compound (MC) data were compared by paired t-test.
P < 0.05,
P < 0.01. AVP, arginine vasopressin.
Similar RBF studies were conducted on CD38−/− mice to determine the importance, if any, of this type of ADP ribosyl cyclase in the acute hemodynamic responses to AVP in the mouse. As is shown in Fig. 2, the maximum responses of RBF, RVR, and MAP to AVP in CD38-null mice (■) did not differ from those in WT mice (○). Thus we conclude that CD38 and its downstream Ca2+ signaling pathway are not involved in the renal vascular actions of AVP mediated by vascular V1a receptor stimulation in the mouse.
Fig. 2.

AVP-induced changes in RBF, RVR, and MAP in WT (○, n = 20) and CD38−/− (■, n = 20) mice. Top: maximum responses produced by AVP (3–25 ng). Bottom: area under curves for the same parameters in WT and CD38−/− mice following AVP injections. There are no differences between WT and CD38−/− mice.
Analysis of the maximum hemodynamic responses to AVP is somewhat limited in that it does not provide insight into the overall magnitude of the response that includes duration as well as the maximum response. To assess this, we calculated the integrated area of the response curves to provide a more comprehensive index of the overall response. These data are summarized in Fig. 2, bottom. The integrated responses of RBF, RVR, and MAP produced by AVP were similar for WT and CD38−/− mice. These results reinforce the conclusion that AVP responses in CD38−/− and WT mice are not different. Because the two approaches to data analysis yielded similar conclusions for group comparisons, subsequent analyses of the magnitude of measured responses are based solely on maximum changes produced by AVP.
Other experiments tested whether NO buffers part of the vasoconstrictor actions of AVP in the mouse. To this end, l-NAME was administered to inhibit NOS in the experimental period. Basal data for these animals are presented in Table 3. l-NAME reduced RBF and increased RVR and MAP in both WT and CD38−/− mice. The maximum vasoconstrictor responses to NOS inhibition were similar in both groups of mice, with changes averaging 50% for RBF, 230% for RVR, and 12% for MAP. Thus resting, tonic formation of NO produces appreciable renal vasodilation that is independent of CD38 under resting conditions. The renal vasoconstrictor actions of AVP were magnified during inhibition of NO production by l-NAME. As is shown in Fig. 3, the reductions in RBF produced by the three lowest doses of AVP were increased more than fourfold during l-NAME treatment, and the highest dose response was increased two- to threefold. The increases in RVR due to AVP were amplified two- to fourfold by NOS inhibition. The systemic pressor response to AVP was less pronounced in the absence of NO. The effect of l-NAME on AVP responses was similar in WT (Fig. 3, top) and CD38−/− mice (Fig. 3, bottom) with no significant differences between the two groups. These results indicate that NO blunts a fraction of AVP-induced renal vasoconstriction and that the contribution of NO is independent of the ADP ribosyl cyclase CD38.
Table 3.
Basal values for l-NAME effects on AVP responses in WT and CD38−/− mice
| WT |
CD38−/− |
|||
|---|---|---|---|---|
| Control | l -NAME | Control | l -NAME | |
| Arterial blood pressure, mmHg | 107 ± 1 | 119 ± 3* | 103 ± 2 | 117 ± 2† |
| Heart rate, beats/min | 474 ± 10 | 391 ± 42 | 428 ± 28 | 379 ± 46 |
| Renal blood flow, ml·min−1·g kidney wt−1 | 6.61 ± 0.44 | 3.32 ± 0.37† | 7.09 ± 0.46 | 3.54 ± 0.26† |
| Renal vascular resistance, mmHg·ml−1·min−1·g kidney wt | 17.1 ± 1.2 | 40.6 ± 4.2† | 15.6 ± 1.0 | 35.7 ± 2.9† |
| Hematocrit, % | 43.4 ± 1.6 | 43.2 ± 2.67 | 40.2 ± 1.4 | 43.0 ± 1.9 |
| Body weight, g | 26.9 ± 0.7 | 25.9 ± 1 | ||
| Right kidney weight, g | 0.190 ± 0.004 | 0.177 ± 0.006 | ||
| Number of animals | 13 | 13 | ||
Values are means ± SE.
P < 0.005,
P < 0.001 for differences between control and N-nitro-l-arginine methyl ester (l-NAME) periods in each strain.
Fig. 3.

Effects of nitric oxide (NO) synthase inhibition with nitro-l-arginine methyl ester (l-NAME) on vascular responses to AVP in WT mice (top: n = 13) and CD38−/− mice (bottom: n = 13). ○, Control responses to AVP (3–25 ng). ■, Responses to AVP after NO synthase inhibition with l-NAME. #P < 0.05 for WT vs. CD38−/− *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001 for difference between control and l-NAME periods.
We next tested for possible buffering of the renal vascular actions of AVP by COX-derived vasodilator prostanoids. Indomethacin was administered in the experimental period to inhibit COX. Indomethacin had a negligible effect on basal renal hemodynamics and MAP in either group of mice (Table 4). COX inhibition magnified the AVP-induced reductions in RBF by 1.5–2.0-fold and the increases in RVR to a lesser extent (Fig. 4), again with no significant differences between responses in WT and CD38−/− mice. The pressor response to AVP was reduced during COX inhibition by ∼30% on the average in both groups of animals. Thus COX-generated vasodilator prostanoids also buffered some of the AVP-induced renal vasoconstriction in a manner independent of CD38 activity.
Table 4.
Basal values for indomethacin effects on AVP responses in WT and CD38−/− mice
| WT |
CD38−/− |
|||
|---|---|---|---|---|
| Control | Indomethacin | Control | Indomethacin | |
| Arterial blood pressure, mmHg | 111 ± 3 | 111 ± 4 | 110 ± 4 | 110 ± 4 |
| Heart rate, beats/min | 466 ± 18 | 400 ± 9* | 459 ± 11 | 392 ± 8† |
| Renal blood flow, ml·min−1·g kidney wt−1 | 11.14 ± 0.63 | 14.10 ± 1.10 | 11.46 ± 0.48 | 13.42 ± 0.93 |
| Renal vascular resistance, mmHg·ml−1·min−1·g kidney wt | 10.4 ± 0.6 | 8.3 ± 0.8 | 10.7 ± 0.6 | 8.5 ± 0.6 |
| Hematocrit, % | 45.1 ± 1.1 | 48.6 ± 1.6 | 40.7 ± 0.7 | 42.1 ± 2.3‡ |
| Body weight, g | 25.5 ± 0.6 | 25.0 ± 0.7 | ||
| Right kidney weight, g | 0.200 ± 0.005 | 0.179 ± 0.006 | ||
| Number of animals | 8 | 7 | ||
Values are means ± SE.
P < 0.01,
P < 0.005 for differences between control and indomethacin period.
P < 0.005 for difference between WT and CD38−/− strain.
Fig. 4.

Effects of cyclooxygenase inhibition with indomethacin on vascular responses to AVP in WT mice (top: n = 8) and CD38−/− mice (bottom: n = 7). ○, control responses to AVP (3–25 ng). ■, Responses to the same doses after indomethacin. There were no differences between CD38−/− and WT mice at any dose during control or indomethacin periods. *P < 0.05, ***P < 0.005, ****P < 0.001 for difference between control and indomethacin treatment in same strain.
In other experiments, we determined the participation of O2·− in AVP-induced renal vasoconstriction by administering tempol in the experimental period to scavenge O2·−. Tempol had a negligible effect on basal renal hemodynamics and MAP in either group of mice (Table 5). As Fig. 5 shows, the antioxidant attenuated the magnitude of the reduction in RBF caused by AVP by 25–50%, whereas the increase in RVR was reduced by 30% in WT and CD38-null mice. Tempol had little effect on the MAP response to AVP in either strain. The similar degree of attenuation by tempol on AVP-mediated vascular responses in WT and CD38−/− mice supports the conclusion that the potentiating action of O2·− is also independent from CD38 and its downstream Ca2+-signaling pathway.
Table 5.
Basal values for tempol effects on AVP responses in WT and CD38−/− mice
| WT |
CD38−/− |
|||
|---|---|---|---|---|
| Control | Tempol | Control | Tempol | |
| Arterial blood pressure, mmHg | 110 ± 3 | 110 ± 3 | 110 ± 2 | 105 ± 3 |
| Heart rate, beats/min | 454 ± 28 | 481 ± 23 | 447 ± 23 | 448 ± 16 |
| Renal blood flow, ml·min−1·g kidney wt−1 | 8.25 ± 0.71 | 10.64 ± 1.11 | 10.76 ± 0.78 | 13.37 ± 1.19 |
| Renal vascular resistance, mmHg·ml−1·min−1·g kidney wt | 15.4 ± 2.2 | 12.0 ± 2.0 | 10.7 ± 0.6 | 8.5 ± 0.9 |
| Hematocrit, % | 42.4 ± 0.9 | 41.6 ± 2.1 | 39.8 ± 1.3 | 45.3 ± 1.2 |
| Body weight, g | 26.5 ± 0.6 | 23.9 ± 0.6 | ||
| Right kidney weight, g | 0.187 ± 0.005 | 0.176 ± 0.007 | ||
| Number of animals | 11 | 10 | ||
Values are means ± SE. There are no significant differences within or between strains.
Fig. 5.

Effects of superoxide quenching with tempol on vascular responses to AVP in WT mice (top: n = 11) and CD38−/− mice (bottom: n = 10). ○, Control responses to AVP (3–25 ng). ■, Responses to the same doses after superoxide inhibition with tempol. #P < 0.05 for WT vs. CD38−/− in control period $P < 0.05 for WT vs CD38−/− after tempol, **P < 0.01, ***P < 0.005, ****P < 0.001 for difference between control and tempol in same strain.
To further investigate the influence of O2·− on vascular responses to AVP, we tested the effects of tempol after NOS inhibition with l-NAME in WT mice. These experiments showed no inhibitory action of tempol after l-NAME treatment; the 25 ng dose of AVP increased the RVR response to 283 ± 81% above control after l-NAME and to 241 ± 96% after tempol combined with l-NAME (NS, n = 9). This demonstrates that O2·− does not contribute to the vasoconstrictor response to AVP in the absence of NO. Our interpretation is that the effect of O2·− on AVP-mediated renal vasoconstriction is primarily due to the neutralization of NO rather than a direct action of O2·− on the excitation-contraction process per se.
Bolus injections of AVP provide transient, dynamic hemodynamic changes with consistent onset and recovery slopes. Response slopes for RBF were directly correlated with the magnitude of the vasoconstriction, and this relationship was used to characterize the dynamics of AVP responses. The rates of decline in RBF following AVP injections (vasoconstrictor response) were not different between the control and experimental periods in any protocol (Fig. 6 and Table 6). Thus, although the magnitude of the vasoconstrictor response to AVP is buffered by NO and prostanoids and augmented by O2·−, these agents do not influence the rate of contraction induced by AVP within the renal microcirculation. Similarly, there were no differences in the rates of vasoconstriction between WT and CD38−/− mice in the control period or during any experimental period (i.e., NO, prostanoid, or O2·− inhibition, Table 6). Pooled values of the vasoconstriction phase provide an overall average rate of vasoconstriction of −0.065 ± 0.004% of basal RBF/s. Unlike the rate of vasoconstriction, the relaxation phase was more complex and strongly influenced by the buffering action of NO and prostanoids. These data are shown in Fig. 6, right, and in Table 6. Both l-NAME and indomethacin slowed the rate of recovery or relaxation from AVP-induced vasoconstriction compared with their respective control responses. This difference was statistically significant for both groups. The rate of relaxation was unaffected by tempol.
Fig. 6.
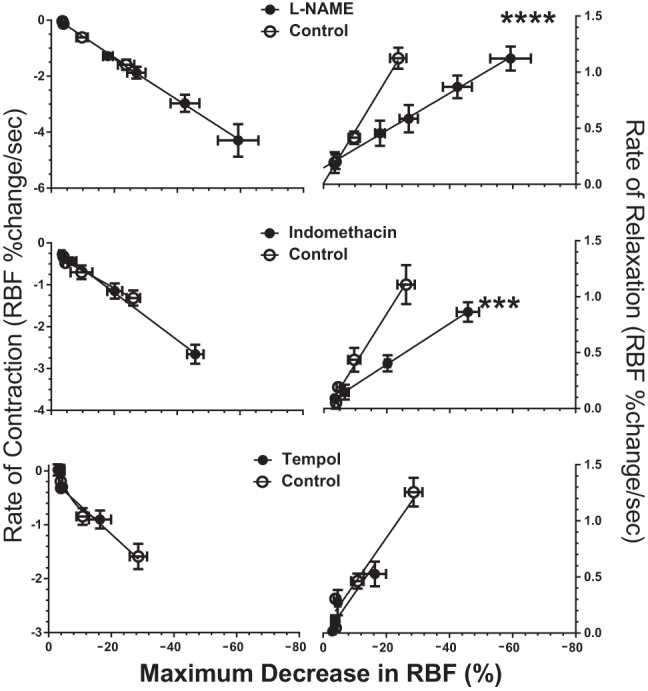
Slopes of RBF changes in WT mice as a function of the maximum reduction in RBF produced by AVP at each dose. Left: rate of constrictions (downslope). Right: rate of relaxations (upslope). Each horizontal pair shows the effects of l-NAME, indomethacin, or tempol. Rate of change in RBF is calculated as percent change from the basal level per second. ***P < 0.005, ****P < 0.001 for difference between control recovery rate and treatment recovery rate.
Table 6.
Rates of contraction (downslope) and relaxation (upslope) as a function of maximum reduction in RBF following AVP injections in WT and CD38−/− mice
| WT |
CD38−/− |
|||||||
|---|---|---|---|---|---|---|---|---|
| Contraction |
Relaxation |
Contraction |
Relaxation |
|||||
| Control | Exp | Control | Exp | Control | Exp | Control | Exp | |
| l-NAME | −75.1 ± 8.4 | −72.7 ± 10.1 | 46.4 ± 4.8 | 16.6 ± 3.3† | −81.3 ± 6.7 | −57.9 ± 9.4 | 33.6 ± 3.9 | 9.1 ± 2.8† |
| Indomethacin | −41.5 ± 7.8 | −56.5 ± 4.4 | 44.9 ± 5.9 | 18.3 ± 2.0* | −54.2 ± 5.8 | −60.5 ± 5.2 | 40.2 ± 4.0 | 6.3 ± 2.1* |
| Tempol | −53.4 ± 7.8 | −66.0 ± 10.7 | 43.7 ± 4.2 | 32.7 ± 7.8 | −67.9 ± 10.4 | −81.5 ± 11.1 | 41.7 ± 4.7 | 33.6 ± 5.5 |
Values are mean % change in basal renal blood flow (RBF)/s maximum reduction in RBF−1 × 1,000 ± SE. Actual slopes are 1/1,000 of values shown.
P < 0.005,
P < 0.001 for difference between control and experimental periods (Exp) in each strain.
The RBF recovery rate after indomethacin treatment was notable as the only slope that showed a difference between CD38−/− and WT mice. The difference was small and of unknown physiological significance. In all other cases, CD38−/− recovery rates were indistinguishable from the corresponding WT values.
DISCUSSION
Our results provide the first assessment of vascular reactivity to AVP in the mouse kidney. We observed that the vascular responses are restricted to vasoconstriction due to AVP V1a receptor activation. We found no evidence for a renal vasodilatory action of AVP at the doses tested, and the AVP V2 receptor agonist dDAVP did not influence RBF or AP. V2 receptor blockade was also ineffective. The magnitude of the V1a receptor-mediated vasoconstriction is modulated significantly by vasodilator systems involving release of NO and COX-generated prostanoids and by the vasoconstrictor actions of reactive oxygen species, probably O2·− anion, as revealed by a quenching action of the superoxide dismutase (SOD) mimetic tempol. The inhibitory action of tempol was eliminated by NOS inactivation with l-NAME, suggesting that the primary role of O2·− on AVP-induced renal vasoconstriction is an indirect one involving reduced availability of NO. A major new finding was that, unlike other vasoconstrictor GPCR agonists tested to date, AVP-induced renal vasoconstriction does not involve the ADP ribosyl cyclase enzyme CD38 (27–29). Moreover, inhibition of O2·− generation with tempol produces the same inhibitory effect on renal vasoconstriction in response to AVP in WT and CD38−/− mice, indicating an action of O2·− that is also independent of CD38. This contrasts with previous reports showing that O2·− induces Ca2+ mobilization and vasoconstriction, in part, by stimulation of cADP ribose production (28, 29, 96, 100). It seems that, unlike other vasoconstrictor GPCRs, the V1a AVP receptor does not engage the CD38/O2·− pathway as part of the contractile response.
Previous studies have provided evidence for predominant V1a receptor mediation of AVP actions on the renal vasculature of the rat (8, 20, 30). V2 receptors in the rat kidney localized to the distal tubule and collecting duct regulate water permeability, but no V2 receptor mRNA or protein expression is found in the rat renal vasculature (20). Consistent with this, pharmacological studies have failed to reveal V2 receptor-mediated renal vasodilation in the rat (8, 30, 57). In contrast, however, AVP activates V2 receptors to produce NO-dependent renal vasodilation in the dog (2, 55, 67) and rabbit (49).
Cyclic ADP ribose in afferent arterioles has emerged as an important component in Ca2+ signaling activated by angiotensin-II (Ang II), endothelin-1 (ET-1), and norepinephrine (NE) (27–29). The direct consequence of this in vivo is evident as a diminished renal vasoconstrictor response to Ang II, ET-1, NE, phenylephrine, and thromboxane in mice lacking the major mammalian ADP ribosyl cyclase CD38 (65, 83–85). These publications raised the possibility that cyclic ADP ribose is utilized as a Ca2+-mobilizing agent by all vasoconstrictor agents that activate Gq/11 protein-coupled receptors in the renal microcirculation. However, we now show that AVP is an exception to this rule. This may be the result of differences in Ca2+ signaling or Ca2+ sensitivity between the excitation/contraction pathways used by AVP and other GPCR vasoconstrictors, an issue worthy of further investigation.
In common with other Gq/11-coupled receptors that activate phospholipase C, the vasoconstrictor action of AVP V1a receptors is mediated in part by Ca2+ entry through l-type Ca2+ channels (9, 25, 26, 32, 42) and mobilization of Ca2+ from internal VSMC stores in response to inositol 1,4,5-trisphosphate (IP3) (32, 81). In vitro studies on afferent arterioles have shown additional Ca2+ entry through store-operated Ca2+ channels triggered by Ca2+ mobilization (26). Several studies into the vasoconstrictor action of AVP on nonrenal arterial VSMC maintained in culture conditions have implicated inhibition of K+ channels (90, 93), resulting in a depolarizing stimulus that promotes Ca2+ entry through voltage-sensitive L-type Ca2+ channels. Others have found that physiological levels of AVP may preferentially inhibit K+ channels and activate transient receptor potential channel 6 (TRPC6) cation channels in mesenteric VSMC independently from the IP3 signaling pathway (38, 59). Such a mechanism independent of Ca2+ mobilization might distinguish the signaling pathways for AVP from other Gq/11-coupled vasoconstrictors.
A strong buffering influence of NO against AVP-mediated vasoconstriction in the mouse kidney is readily apparent from the marked potentiating effect of l-NAME inhibition of NOS. Early studies on isolated-perfused kidneys suggested that NO buffering of AVP-mediated renal vasoconstriction is the result of endothelial NOS activation by increased shear stress (8). However, in vivo vasoconstrictor responses to Ang II in the rat (23, 45) and the rabbit (74) and thromboxane agonists in the mouse (N. Moss and W, Arendshorst, unpublished observations) are weakly or not at all potentiated by l-NAME, suggesting that V1a receptor-mediated AVP-induced vasoconstriction is more actively buffered by NO compared with other vasoconstrictor agents. This is supported by a direct renal vasodilatory response to subpressor doses of AVP in rats that is inhibited by l-NAME (75). Other reports have shown that selective activation of V1 receptors in the rat promote production of NO and vasodilator prostaglandins (PGs) in the renal vasculature that offset the vasoconstrictor action of AVP (19, 57). This is not limited to V1 receptors, as NO-dependent vasodilatation is stimulated by V2 receptor activation in the dog (2, 55, 67) and rabbit (82). In the present study, we observed no vasodilatory responses to AVP. The lowest tested dose of AVP reduced RBF by 3%, increased RVR by 15%, and increased MAP by 10–20%. Our data in the mouse support direct stimulation of NO production by AVP to buffer the vasoconstrictor response involving V1a, but not V2, receptor activation.
In addition to l-NAME, COX inhibition magnified AVP-induced renal vasoconstriction, implicating primary inhibition of production of a vasodilatory PG, such as PGE2 and/or PGI2. Several reports have implicated PGs as a buffering agent against the systemic and renal vascular response to AVP in the rat (19, 30, 39, 94), dog (70), rabbit (41, 76), and humans (66). We cannot exclude the possibility that synthesis of the vasoconstrictor thromboxane (TxA) was also attenuated, but previous studies have shown that basal production of TxA2 is quite low in healthy kidneys, and it normally exerts little basal renal vasoconstriction in vivo (10, 12, 35, 44, 89). Moreover, intrarenal infusion of AVP causes COX-dependent release of PGE2 and PGI2 (19, 58, 63) but not TxA2 (63). Overall, the net effect of COX inhibition in our mice was removal of vasodilator prostanoids that normally buffer renal vasoconstriction produced by AVP.
Superoxide anions have not been shown previously to participate in AVP-mediated vasoconstriction in the kidney. The potentiating role of O2·− in renal vasoconstrictor responses to angiotensin AT1 and thromboxane prostanoid receptor activation are well documented (47, 65, 73), but little is known for AVP. Previous studies on the interactions between AVP and O2·− in nonrenal arteries and VSMC have shown upregulation of the V1a receptor by O2·− (52) and an action of AVP to stimulate O2·− production (54). AVP stimulation of O2·− production by NADPH oxidase is reported to increase TRPC6 channel activity and Ca2+ entry in the A7r5 line of cultured VSMC and mouse aortic VSMC (24). In our study, a potentiating action of O2·− on the renal vasoconstrictor response to AVP is clearly supported by an inhibitory action of the SOD mimetic tempol on the vascular response, but the mechanisms responsible for this action of O2·− are uncertain. Possibilities include the suppression of NO-mediated buffering due to the mutually destructive reaction between O2·− and NO and a direct vasoconstrictor action of O2·− through activation of Ca2+ signaling pathways. The lack of effect of tempol treatment after NOS inhibition provides insight into the relative importance of these two possibilities. The effect of O2·− to reduce NO availability would be nullified by prior inhibition of NO production and its reduced bioavailability, whereas a direct action of O2·− on the contractile response of VSMC would be expected to persist after l-NAME. Because the inhibitory action of tempol on vascular activity of AVP was not evident after NOS inhibition with l-NAME, we conclude that the principle potentiating effect of O2·− is to reduce the availability of NO. This also implies that O2·− generated by AVP does not participate in cellular Ca2+ signaling, such as the ADP ribosyl cyclase/RyR/Ca2+ pathway (28, 29). The similarity between renal vasoconstrictor responses to AVP in WT and CD38−/− mice is quite consistent with this conclusion and represents a clear distinction between AVP V1a receptors and other vasoconstrictor GPCRs. This could result in different susceptibilities to the effects of oxidative stress and inflammatory reactions during the development of disease states.
Analysis of dynamic components in the vasoconstrictor and relaxation responses to AVP is a novel approach to characterize acute, transient RBF responses. Our data indicate that, after AVP injection, RBF declined at a rate that was directly proportional to the maximal reduction in RBF. Interestingly, this rate of vasoconstriction was not different between WT and CD38−/− animals and was unaffected by treatment with l-NAME, indomethacin, or tempol. Future studies are required to determine whether the constancy of this relationship is a function of the intrinsic contractility of the renal vasculature or whether it varies with different vasoconstrictor agents and their respective GPCR. In contrast, the relaxation phase following AVP-mediated constriction was clearly prolonged by l-NAME and indomethacin. This is readily explained by the suppression of vasodilatory stimuli from NO and prostanoids that would normally accelerate the rate of relaxation of VSMC. Previous studies in the rat have shown that PGE2 and PGI2 and their analogs accelerate the rate of relaxation following Ang II-, norepinephrine-, and TxA2-induced renal vasoconstriction (18). It is notable that the relaxation rate was slowed to the same extent by l-NAME and indomethacin, even though the RVR response to the highest dose of AVP was increased more by l-NAME (308%) than indomethacin (125%). This is consistent with the notion that NO and prostanoids engage a similar vasodilatory mechanism to buffer the vasoconstrictor effects of AVP. Unlike vasodilator inhibition, suppression of O2·− with tempol did not alter the recovery rate from AVP-induced vasoconstriction. The extremely short half-life of O2·− may limit its biological activity to the contractile phase of the vascular response.
Our new information documents that AVP is a potent renal vasoconstrictor in the mouse, an action that is normally strongly counteracted in vivo by vasodilatory actions of NO and prostanoids. A modest increase in RBF and decrease in RVR following inhibition of V1a receptor activity indicate a small tonic vasoconstrictor effect of circulating endogenous AVP on the renal circulation. Previous studies have reported that physiological plasma concentrations of AVP (<10 pg/ml) or V1 receptor antagonism does not appreciably affect MAP, RBF, or RVR in conscious or pentobarbital-anesthetized rats (15, 34, 36, 48, 68) or conscious dogs (16). Perhaps endogenous AVP has weaker renal vasoconstrictor actions in other species in which buffering of vasoconstriction by vasodilator systems involving NO and prostanoids exert a larger effect. On the other hand, V1a antagonists can produce a systemic hypotensive effect during severe dehydration in most species when circulating AVP levels approach maximum endogenous levels (13, 95). Similarly, vasopressor and renal vasoconstrictor effects of exogenous AVP administered at more physiological levels are typically more readily apparent when normal buffering or offsetting vasodilator mechanisms are suppressed experimentally (94) or in pathological states such as hypertension (17, 64, 77, 98). AVP produces exaggerated Ca2+ signaling and vasoconstriction of renal microvessels of SHR with genetic hypertension (30, 32, 43, 88), largely due to upregulation of V1a receptor mRNA and protein levels (87). These models likely represent situations when plasma AVP is elevated and vasodilatory buffering actions are suppressed, most notably by oxidative stress and excessive O2·− levels in VSMC (5).
Under normal circumstances, the renal medullary microcirculation is more responsive than the cortical circulation to changes in plasma AVP concentration, with a sensitivity that parallels that of the collecting duct (20, 33). In normal rats, localized AVP infusions into the renal medulla to activate V1a receptors results in increased AP when not offset by NO secondary to V2 receptor activation (21, 79). Nevertheless, from a systemic standpoint, the mesenteric and hindquarter circulations appear to be more sensitive to AVP than the renal microvasculature (14, 36, 40, 97) perhaps due to more effective buffering of the renal vasomotor actions of AVP. This may contribute to better maintenance of RBF and protection of kidney function when AVP is compared with other vasoconstrictor agents used to support blood pressure during hemorrhagic or septic shock (3, 46, 53).
GRANTS
This work was supported by NIH grant RO1 HL-02334.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: N.G.M. and W.J.A. conception and design of research; N.G.M. and T.E.K. performed experiments; N.G.M. analyzed data; N.G.M. and W.J.A. interpreted results of experiments; N.G.M. prepared figures; N.G.M. and W.J.A. drafted manuscript; N.G.M. and W.J.A. edited and revised manuscript; N.G.M., T.E.K., and W.J.A. approved final version of manuscript.
REFERENCES
- 1.Ackermann U, Azizi N. Increased central AT(1)-receptor activation, not systemic vasopressin, sustains hypertension in ANP knockout mice. Am J Physiol Regul Integr Comp Physiol 278: R1441–R1445, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Aki Y, Tamaki T, Kiyomoto H, He H, Yoshida H, Iwao H, Abe Y. Nitric oxide may participate in V2 vasopressin-receptor-mediated renal vasodilation. J Cardiovasc Pharmacol 23: 331–336, 1994 [PubMed] [Google Scholar]
- 3.Anand T, Skinner R. Arginine vasopressin: the future of pressure-support resuscitation in hemorrhagic shock. J Surg Res 178: 321–329, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Aoyagi T, Koshimizu TA, Tanoue A. Vasopressin regulation of blood pressure and volume: findings from V1a receptor-deficient mice. Kidney Int 76: 1035–1039, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Araujo M, Wilcox CS. Oxidative stress in hypertension: role of the kidney. Antioxid Redox Signal 20: 74–101, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakris G, Bursztyn M, Gavras I, Bresnahan M, Gavras H. Role of vasopressin in essential hypertension: racial differences. J Hypertens 15: 545–550, 1997 [DOI] [PubMed] [Google Scholar]
- 8.Barthelmebs M, Krieger JP, Grima M, Nisato D, Imbs JL. Vascular effects of [Arg8]vasopressin in the isolated perfused rat kidney. Eur J Pharmacol 314: 325–332, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Bauer J, Parekh N. Variations in cell signaling pathways for different vasoconstrictor agonists in renal circulation of the rat. Kidney Int 63: 2178–2186, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Baylis C, Slangen B, Hussain S, Weaver C. Relationship between basal NO release and cyclooxygenase products in the normal rat kidney. Am J Physiol Regul Integr Comp Physiol 271: R1327–R1334, 1996 [DOI] [PubMed] [Google Scholar]
- 11.Berecek KH, Schwertschlag U, Gross F. Alterations in renal vascular resistance and reactivity in spontaneous hypertension of rats. Am J Physiol Heart Circ Physiol 238: H287–H293, 1980 [DOI] [PubMed] [Google Scholar]
- 12.Boffa JJ, Just A, Coffman TM, Arendshorst WJ. Thromboxane receptor mediates renal vasoconstriction and contributes to acute renal failure in endotoxemic mice. J Am Soc Nephrol 15: 2358–2365, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Brand PH, Metting PJ, Britton SL. Support of arterial blood pressure by major pressor systems in conscious dogs. Am J Physiol Heart Circ Physiol 255: H483–H491, 1988 [DOI] [PubMed] [Google Scholar]
- 14.Brizzee BL, Harrison-Bernard L, Pretus HA, Clifton GG, Walker BR. Hemodynamic responses to vasopressinergic antagonism in water-deprived conscious rats. Am J Physiol Regul Integr Comp Physiol 255: R46–R51, 1988 [DOI] [PubMed] [Google Scholar]
- 15.Brizzee BL, Walker BR. Cardiovascular responses to V1-vasopressinergic antagonism in conscious versus anesthetized rats. Can J Physiol Pharmacol 66: 1437–1441, 1988 [DOI] [PubMed] [Google Scholar]
- 16.Brown AJ, Lohmeier TE, Carroll RG, Meydrech EF. Cardiovascular and renal responses to chronic vasopressin infusion. Am J Physiol Heart Circ Physiol 250: H584–H594, 1986 [DOI] [PubMed] [Google Scholar]
- 17.Burrell LM, Phillips PA, Risvanis J, Aldred KL, Hutchins AM, Johnston CI. Attenuation of genetic hypertension after short-term vasopressin V1A receptor antagonism. Hypertension 26: 828–834, 1995 [DOI] [PubMed] [Google Scholar]
- 18.Chatziantoniou C, Arendshorst WJ. Prostaglandin interactions with angiotensin, norepinephrine, and thromboxane in rat renal vasculature. Am J Physiol Renal Fluid Electrolyte Physiol 262: F68–F76, 1992 [DOI] [PubMed] [Google Scholar]
- 19.Cooper CL, Malik KU. Mechanism of action of vasopressin on prostaglandin synthesis and vascular function in the isolated rat kidney: effect of calcium antagonists and calmodulin inhibitors. J Pharmacol Exp Ther 229: 139–147, 1984 [PubMed] [Google Scholar]
- 20.Cowley AW., Jr Control of the renal medullary circulation by vasopressin V1 and V2 receptors in the rat. Exp Physiol 85: 223S–231S, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Cowley AW, Jr, Skelton MM, Kurth TM. Effects of long-term vasopressin receptor stimulation on medullary blood flow and arterial pressure. Am J Physiol Regul Integr Comp Physiol 275: R1420–R1424, 1998 [DOI] [PubMed] [Google Scholar]
- 22.Cowley AW, Jr, Szczepanska-Sadowska E, Stepniakowski K, Mattson D. Chronic intravenous administration of V1 arginine vasopressin agonist results in sustained hypertension. Am J Physiol Heart Circ Physiol 267: H751–H756, 1994 [DOI] [PubMed] [Google Scholar]
- 23.Dautzenberg M, Just A. Temporal characteristics of nitric oxide-, prostaglandin-, and EDHF-mediated components of endothelium-dependent vasodilation in the kidney. Am J Physiol Regul Integr Comp Physiol 305: R987–R998, 2013 [DOI] [PubMed] [Google Scholar]
- 24.Ding Y, Winters A, Ding M, Graham S, Akopova I, Muallem S, Wang Y, Hong JH, Gryczynski Z, Yang SH, Birnbaumer L, Ma R. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J Biol Chem 286: 31799–31809, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fallet RW, Ikenaga H, Bast JP, Carmines PK. Relative contributions of Ca2+ mobilization and influx in renal arteriolar contractile responses to arginine vasopressin. Am J Physiol Renal Physiol 288: F545–F551, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fellner SK, Arendshorst WJ. Capacitative calcium entry in smooth muscle cells from preglomerular vessels. Am J Physiol Renal Physiol 277: F533–F542, 1999 [DOI] [PubMed] [Google Scholar]
- 27.Fellner SK, Arendshorst WJ. Angiotensin II Ca2+ signaling in rat afferent arterioles: stimulation of cyclic ADP ribose and IP3 pathways. Am J Physiol Renal Physiol 288: F785–F791, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Fellner SK, Arendshorst WJ. Angiotensin II, reactive oxygen species and Ca2+ signaling in afferent arterioles. Am J Physiol Renal Physiol 289: F1012–F1019, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Fellner SK, Arendshorst WJ. Endothelin-A and -B receptors, superoxide, and Ca2+ signaling in afferent arterioles. Am J Physiol Renal Physiol 292: F175–F184, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Feng JJ, Arendshorst WJ. Enhanced renal vasoconstriction induced by vasopressin in SHR is mediated by V1 receptors. Am J Physiol Renal Fluid Electrolyte Physiol 271: F304–F313, 1996 [DOI] [PubMed] [Google Scholar]
- 32.Feng JJ, Arendshorst WJ. Calcium signaling mechanisms in renal vascular responses to vasopressin in genetic hypertension. Hypertension 30: 1223–1231, 1997 [DOI] [PubMed] [Google Scholar]
- 33.Franchini KG, Cowley AW., Jr Renal cortical and medullary blood flow responses during water restriction: role of vasopressin. Am J Physiol Regul Integr Comp Physiol 270: R1257–R1264, 1996 [DOI] [PubMed] [Google Scholar]
- 34.Gellai M, Silverstein JH, Hwang JC, LaRochelle FT, Jr, Valtin H. Influence of vasopressin on renal hemodynamics in conscious Brattleboro rats. Am J Physiol Renal Fluid Electrolyte Physiol 246: F819–F827, 1984 [DOI] [PubMed] [Google Scholar]
- 35.Grone HJ, Grippo RS, Arendshorst WJ, Dunn MJ. Role of thromboxane in control of arterial pressure and renal function in young spontaneously hypertensive rats. Am J Physiol Renal Fluid Electrolyte Physiol 250: F488–F496, 1986 [DOI] [PubMed] [Google Scholar]
- 36.Harrison-Bernard LM, Brizzee BL, Clifton GG, Walker BR. Renal versus hindquarter hemodynamic responses to vasopressin in conscious rats. J Cardiovasc Pharmacol 16: 719–726, 1990 [DOI] [PubMed] [Google Scholar]
- 37.Hasser EM, Bishop VS, Hay M. Interactions between vasopressin and baroreflex control of the sympathetic nervous system. Clin Exp Pharmacol Physiol 24: 102–108, 1997 [DOI] [PubMed] [Google Scholar]
- 38.Henderson KK, Byron KL. Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J Appl Physiol 102: 1402–1409, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hofbauer KG, Dienemann H, Forgiarini P, Stalder R, Wood JM. Renal vascular effects of angiotensin II, arginine-vasopressin and bradykinin in rats: interactions with prostaglandins. Gen Pharmacol 14: 145–147, 1983 [DOI] [PubMed] [Google Scholar]
- 40.Hofbauer KG, Studer W, Mah SC, Michel JB, Wood JM, Stalder R. The significance of vasopressin as a pressor agent. J Cardiovasc Pharmacol 6, Suppl 2: S429–S438, 1984 [DOI] [PubMed] [Google Scholar]
- 41.Inaba M, Katayama S, Itabashi A, Maruno Y, Ishii J. Effects of arginine vasopressin on blood pressure and renal prostaglandin E2 in rabbits. Endocrinol Jpn 38: 505–509, 1991 [DOI] [PubMed] [Google Scholar]
- 42.Iversen BM, Arendshorst WJ. ANG II and vasopressin stimulate calcium entry in dispersed smooth muscle cells of preglomerular arterioles. Am J Physiol Renal Physiol 274: F498–F508, 1998 [DOI] [PubMed] [Google Scholar]
- 43.Iversen BM, Arendshorst WJ. Exaggerated Ca2+ signaling in preglomerular arteriolar smooth muscle cells of genetically hypertensive rats. Am J Physiol Renal Physiol 276: F260–F270, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Jackson EK, Goto F, Uderman HD, Workman RJ, Herzer WA, FitzGerald GA, Branch RA. Effects of thromboxane synthase inhibitors on renal function. Naunyn Schmiedebergs Arch Pharmacol 337: 183–190, 1988 [DOI] [PubMed] [Google Scholar]
- 45.Just A, Olson AJ, Whitten CL, Arendshorst WJ. Superoxide mediates acute renal vasoconstriction produced by angiotensin II and catecholamines by a mechanism independent of nitric oxide. Am J Physiol Heart Circ Physiol 292: H83–H92, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Kampmeier TG, Rehberg S, Westphal M, Lange M. Vasopressin in sepsis and septic shock. Minerva Anestesiol 76: 844–850, 2010 [PubMed] [Google Scholar]
- 47.Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol 13: 2860–2868, 2002 [DOI] [PubMed] [Google Scholar]
- 48.Keen HL, Sigmund CD. Paradoxical regulation of short promoter human renin transgene by angiotensin ii. Hypertension 37: 403–407, 2001 [DOI] [PubMed] [Google Scholar]
- 49.Kiyomoto K, Tamaki T, Tomohiro A, Nishiyama A, Aki Y, Kimura S, Abe Y. Role of nitric oxide in desmopressin-induced vasodilation of microperfused rabbit afferent arterioles. Hypertens Res 20: 29–34, 1997 [DOI] [PubMed] [Google Scholar]
- 50.Knepper MA. Molecular physiology of urinary concentrating mechanism: regulation of aquaporin water channels by vasopressin. Am J Physiol Renal Physiol 272: F3–F12, 1997 [DOI] [PubMed] [Google Scholar]
- 51.Koshimizu TA, Nakamura K, Egashira N, Hiroyama M, Nonoguchi H, Tanoue A. Vasopressin V1a and V1b receptors: from molecules to physiological systems. Physiol Rev 92: 1813–1864, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Krauskopf A, Lhote P, Petermann O, Ruegg UT, Buetler TM. Cyclosporin A generates superoxide in smooth muscle cells. Free Radic Res 39: 913–919, 2005 [DOI] [PubMed] [Google Scholar]
- 53.Laporte R, Kohan A, Heitzmann J, Wisniewska H, Toy J, La E, Tariga H, Alagarsamy S, Ly B, Dykert J, Qi S, Wisniewski K, Galyean R, Croston G, Schteingart CD, Riviere PJ. Pharmacological characterization of FE 202158, a novel, potent, selective, and short-acting peptidic vasopressin V1a receptor full agonist for the treatment of vasodilatory hypotension. J Pharmacol Exp Ther 337: 786–796, 2011 [DOI] [PubMed] [Google Scholar]
- 54.Li L, Galligan JJ, Fink GD, Chen AF. Vasopressin induces vascular superoxide via endothelin-1 in mineralocorticoid hypertension. Hypertension 41: 663–668, 2003 [DOI] [PubMed] [Google Scholar]
- 55.Liard JF. L-NAME antagonizes vasopressin V2-induced vasodilatation in dogs. Am J Physiol Heart Circ Physiol 266: H99–H106, 1994 [DOI] [PubMed] [Google Scholar]
- 57.Loichot C, Cazaubon C, De Jong W, Helwig JJ, Nisato D, Imbs JL, Barthelmebs M. Nitric oxide, but not vasopressin V2 receptor-mediated vasodilation, modulates vasopressin-induced renal vasoconstriction in rats. Naunyn Schmiedebergs Arch Pharmacol 361: 319–326, 2000 [DOI] [PubMed] [Google Scholar]
- 58.Lote CJ, Thewles A, Wood JA. Vasopressin-induced natriuresis in the conscious rat: role of blood pressure, renal prostaglandin synthesis and the peptide ANF. J Physiol 411: 481–491, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, Byron KL. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther 325: 475–483, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Majid DSA, Navar LG. Nitric oxide in the control of renal hemodynamics and excretory function. Am J Hypertens 14: 74S–82S, 2001 [DOI] [PubMed] [Google Scholar]
- 61.Majid DSA, Said KE, Omoro SA, Navar LG. Nitric oxide dependency of arterial pressure-induced changes in renal interstitial hydrostatic pressure in dogs. Circ Res 88: 347–351, 2001 [DOI] [PubMed] [Google Scholar]
- 62.Manning M, Przybylski JP, Olma A, Klis WA, Kruszynski M, Wo NC, Pelton GH, Sawyer WH. No requirements of cyclic conformation of antagonists in binding to vasopressin receptors. Nature 329: 839–840, 1987 [DOI] [PubMed] [Google Scholar]
- 63.Miller MJ, Carrara MC, Westlin WF, McNeill H, McGiff JC. Compartmental prostaglandin release by angiotensin II and arginine-vasopressin in rabbit isolated perfused kidneys. Eur J Pharmacol 120: 43–50, 1986 [DOI] [PubMed] [Google Scholar]
- 64.Mohring J, Kintz J, Schoun J. Studies on the role of vasopressin in blood pressure control of spontaneously hypertensive rats with established hypertension (SHR, stroke-prone strain). J Cardiovasc Pharmacol 1: 593–608, 1979 [DOI] [PubMed] [Google Scholar]
- 65.Moss NG, Vogel PA, Kopple TE, Arendshorst WJ. Thromboxane-induced renal vasoconstriction is mediated by the ADP-ribosyl cyclase CD38 and superoxide anion. Am J Physiol Renal Physiol 305: F830–F838, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nadler J, Zipser RD, Coleman R, Horton R. Stimulation of renal prostaglandins by pressor hormones in man: comparison of prostaglandin E2 and prostacyclin (6 keto prostaglandin F1 alpha). J Clin Endocrinol Metab 56: 1260–1265, 1983 [DOI] [PubMed] [Google Scholar]
- 67.Naitoh M, Suzuki N, Murakami M, Matsumoto A, Ichihara A, Nakamoto H, Yamamura Y, Saruta T. Arginine vasopressin produces renal vasodilation via V2 receptors in conscious dogs. Am J Physiol Regul Integr Comp Physiol 265: R934–R942, 1993 [DOI] [PubMed] [Google Scholar]
- 68.Nakamura T, Sakamaki T, Kurashina T, Hoshino J, Sato K, Ono Z, Murata K. Effect of vasopressin V1 (OPC-21268) and V2 (OPC-31260) antagonists on renal hemodynamics and excretory function. Life Sci 55: PL67–PL72, 1994 [DOI] [PubMed] [Google Scholar]
- 69.Oikawa R, Hosoda C, Nasa Y, Daicho T, Tanoue A, Tsujimoto G, Takagi N, Tanonaka K, Takeo S. Decreased susceptibility to salt-induced hypertension in subtotally nephrectomized mice lacking the vasopressin V1a receptor. Cardiovasc Res 87: 187–194, 2010 [DOI] [PubMed] [Google Scholar]
- 70.Oliver JA, Sciacca RR, Le CG, Cannon PJ. Modulation by prostaglandins of the renal vascular action of arginine vasopressin. Prostaglandins 24: 641–656, 1982 [DOI] [PubMed] [Google Scholar]
- 71.Patzak A, Lai EY, Mrowka R, Steege A, Persson PB, Persson AE. AT1 receptors mediate angiotensin II-induced release of nitric oxide in afferent arterioles. Kidney Int 66: 1949–1958, 2004 [DOI] [PubMed] [Google Scholar]
- 72.Perucca J, Bichet DG, Bardoux P, Bouby N, Bankir L. Sodium excretion in response to vasopressin and selective vasopressin receptor antagonists. J Am Soc Nephrol 19: 1721–1731, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pfister SL, Nithipatikom K, Campbell WB. Role of superoxide and thromboxane receptors in acute angiotensin II-induced vasoconstriction of rabbit vessels. Am J Physiol Heart Circ Physiol 300: H2064–H2071, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rajapakse NW, Oliver JJ, Evans RG. Nitric oxide in responses of regional kidney blood flow to vasoactive agents in anesthetized rabbits. J Cardiovasc Pharmacol 40: 210–219, 2002 [DOI] [PubMed] [Google Scholar]
- 75.Rudichenko VM, Beierwaltes WH. Arginine vasopressin-induced renal vasodilation mediated by nitric oxide. J Vasc Res 32: 100–105, 1995 [DOI] [PubMed] [Google Scholar]
- 76.Seino M, Abe K, Tsunoda K, Yoshinaga K. Interaction of vasopressin and prostaglandins through calcium ion in the renal circulation. Hypertension 7: 53–58, 1985 [DOI] [PubMed] [Google Scholar]
- 77.Sladek CD, Blair ML, Sterling C, Mangiapane ML. Attenuation of spontaneous hypertension in rats by a vasopressin antagonist. Hypertension 12: 506–512, 1988 [DOI] [PubMed] [Google Scholar]
- 78.Stockand JD. The role of the epithelial Na(+) channel (ENaC) in high AVP but low aldosterone states. Front Physiol 3: 304, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Szczepanska-Sadowska E, Stepniakowski K, Skelton MM, Cowley AW., Jr Prolonged stimulation of intrarenal V1 vasopressin receptors results in sustained hypertension. Am J Physiol Regul Integr Comp Physiol 267: R1217–R1225, 1994 [DOI] [PubMed] [Google Scholar]
- 80.Szentivanyi M, Jr, Park F, Maeda CY, Cowley AW., Jr Nitric oxide in the renal medulla protects from vasopressin-induced hypertension. Hypertension 35: 740–745, 2000 [DOI] [PubMed] [Google Scholar]
- 81.Takahara A, Suzuki-Kusaba M, Hisa H, Satoh S. Effects of a novel Ca2+ entry blocker, CD-349, and TMB-8 on renal vasoconstriction induced by angiotensin II and vasopressin in dogs. J Cardiovasc Pharmacol 16: 966–970, 1990 [DOI] [PubMed] [Google Scholar]
- 82.Tamaki T, Kiyomoto K, He H, Tomohiro A, Nishiyama A, Aki Y, Kimura S, Abe Y. Vasodilation induced by vasopressin V2 receptor stimulation in afferent arterioles. Kidney Int 49: 722–729, 1996 [DOI] [PubMed] [Google Scholar]
- 83.Thai TL, Arendshorst WJ. ADP-ribosyl cyclase and ryanodine receptors mediate endothelin ETA and ETB receptor-induced renal vasoconstriction in vivo. Am J Physiol Renal Physiol 295: F360–F368, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thai TL, Arendshorst WJ. Mice lacking the ADP ribosyl cyclase CD38 exhibit attenauated renal vasoconstriction to angiotensin II, endothelin-1, and norepinephrine. Am J Physiol Renal Physiol 297: F169–F176, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thai TL, Fellner SK, Arendshorst WJ. ADP-ribosyl cyclase and ryanodine receptor activity contribute to basal renal vasomotor tone and agonist-induced renal vasoconstriction in vivo. Am J Physiol Renal Physiol 293: F1107–F1114, 2007 [DOI] [PubMed] [Google Scholar]
- 86.Turner MR, Pallone TL. Vasopressin constricts outer medullary descending vasa recta isolated from rat kidneys. Am J Physiol Renal Physiol 272: F147–F151, 1997 [DOI] [PubMed] [Google Scholar]
- 87.Vagnes OB, Feng JJ, Iversen BM, Arendshorst WJ. Upregulation of V1 receptors in renal resistance vessels of rats developing genetic hypertension. Am J Physiol Renal Physiol 278: F940–F948, 2000 [DOI] [PubMed] [Google Scholar]
- 88.Vagnes OB, Hansen FH, Feng JJ, Iversen BM, Arendshorst WJ. Enhanced Ca2+ response to AVP in preglomerular vessels from rats with genetic hypertension during different hydration states. Am J Physiol Renal Physiol 288: F1249–F1256, 2005 [DOI] [PubMed] [Google Scholar]
- 89.Vagnes OB, Iversen BM, Arendshorst WJ. Short-term ANG II produces renal vasoconstriction independent of TP receptor activation and TxA2/isoprostane production. Am J Physiol Renal Physiol 293: F860–F867, 2007 [DOI] [PubMed] [Google Scholar]
- 90.Van Renterghem C, Romey G, Lazdunski M. Vasopressin modulates the electrical activity in aortic cells (A7r5) by acting on three different types of ionic channels. Proc Natl Acad Sci USA 85: 9365–9369, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vavra I, Machova A, Krejci I. Antidiuretic action of 1-deamino-(8-D-arginine)-vasopressin in unanesthetized rats. J Pharmacol Exp Ther 188: 241–247, 1974 [PubMed] [Google Scholar]
- 92.Viegas VU, Liu ZZ, Nikitina T, Perlewitz A, Zavaritskaya O, Schlichting J, PErsson PB, Regitz-Zagrosek V, Patzak A, Sendeski MM. Angiotensin II type 2 receptor mediates sex differences in mice renal interlobar arteries response to angiotensin II. J Hypertens 30: 1791–1798, 2012 [DOI] [PubMed] [Google Scholar]
- 93.Wakatsuki T, Nakaya Y, Inoue I. Vasopressin modulates K+-channel activities of cultured smooth muscle cells from porcine coronary artery. Am J Physiol Heart Circ Physiol 263: H491–H496, 1992 [DOI] [PubMed] [Google Scholar]
- 94.Walker BR, Brizzee BL, Harrison-Bernard LM. Potentiated vasoconstrictor response to vasopressin following meclofenamate in conscious rats. Proc Soc Exp Biol Med 187: 157–164, 1988 [DOI] [PubMed] [Google Scholar]
- 95.Woods RL, Johnston CI. Contribution of vasopressin to the maintenance of blood pressure during dehydration. Am J Physiol Renal Fluid Electrolyte Physiol 245: F615–F621, 1983 [DOI] [PubMed] [Google Scholar]
- 96.Xu M, Li XX, Ritter JK, Abais JM, Zhang Y, Li PL. Contribution of NADPH oxidase to membrane CD38 internalization and activation in coronary arterial myocytes. PLoS One 8: e71212, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yamada H, Yukimura T, Ito K, Takenaga T, Yamashita Y, Miura K, Yamamoto K. Renal effects of specific antagonists of arginine vasopressin in dogs. Jpn J Pharmacol 55: 19–25, 1991 [DOI] [PubMed] [Google Scholar]
- 98.Yamada Y, Yamamura Y, Chihara T, Onogawa T, Nakamura S, Yamashita T, Mori T, Tominaga M, Yabuuchi Y. OPC-21268, a vasopressin V1 antagonist, produces hypotension in spontaneously hypertensive rats. Hypertension 23: 200–204, 1994 [DOI] [PubMed] [Google Scholar]
- 99.Zhang AY, Chen YF, Zhang DX, Yi FX, Qi J, ndrade-Gordon P, de GL, Li PL, Zou AP. Urotensin II is a nitric oxide-dependent vasodilator and natriuretic peptide in the rat kidney. Am J Physiol Renal Physiol 285: F792–F798, 2003 [DOI] [PubMed] [Google Scholar]
- 100.Zhang AY, Yi F, Teggatz EG, Zou AP, Li PL. Enhanced production and action of cyclic ADP-ribose during oxidative stress in small bovine coronary arterial smooth muscle. Microvasc Res 67: 159–167, 2004 [DOI] [PubMed] [Google Scholar]
- 101.Zhang Z, Rhinehart K, Solis G, Pittner J, Lee-Kwon W, Welch WJ, Wilcox CS, Pallone TL. Chronic ANG II infusion increases NO generation by rat descending vasa recta. Am J Physiol Heart Circ Physiol 288: H29–H36, 2005 [DOI] [PubMed] [Google Scholar]