

Abstract
Helicobacter pylori (H. pylori) infection is one of the most common infections in human beings worldwide. H. pylori express lipopolysaccharides and flagellin that do not activate efficiently Toll-like receptors and express dedicated effectors, such as γ-glutamyl transpeptidase, vacuolating cytotoxin (vacA), arginase, that actively induce tolerogenic signals. In this perspective, H. pylori can be considered as a commensal bacteria belonging to the stomach microbiota. However, when present in the stomach, H. pylori reduce the overall diversity of the gastric microbiota and promote gastric inflammation by inducing Nod1-dependent pro-inflammatory program and by activating neutrophils through the production of a neutrophil activating protein. The maintenance of a chronic inflammation in the gastric mucosa and the direct action of virulence factors (vacA and cytotoxin-associated gene A) confer pro-carcinogenic activities to H. pylori. Hence, H. pylori cannot be considered as symbiotic bacteria but rather as part of the pathobiont. The development of a H. pylori vaccine will bring health benefits for individuals infected with antibiotic resistant H. pylori strains and population of underdeveloped countries.
Keywords: Helicobacter pylori, Vaccine, Immune response, Peptic ulcer, Gastric cancer
Core tip: Helicobacter pylori (H. pylori) infection is one of the most common infections in human beings worldwide. H. pylori actively induce tolerogenic signals and can be considered as a commensal bacteria belonging to the stomach microbiota. However, H. pylori also promote a chronic inflammation in the gastric mucosa and the direct action of virulence factors confers pro-carcinogenic activities to H. pylori. Hence, H. pylori cannot be considered as symbiotic bacteria but rather as part of the pathobiont. The development of a H. pylori vaccine will bring health benefits for individuals infected with antibiotic resistant H. pylori strains and population of underdeveloped countries.
INTRODUCTION
Helicobacter pylori (H. pylori) infection is one of the most common infections in human beings worldwide[1]. After entering the stomach, this spiral, Gram-negative, micro-aerophilic bacterium penetrates the mucus gastric layer[2] but does not traverse the epithelial barrier[3], and therefore it is considered as a non-invasive bacteria. Most of H. pylori organisms are free living in the mucus layer, but some organisms attach to the apical surface of gastric epithelial cells[3] and small numbers have been shown to invade epithelial cells[4]. Humans carry an estimated of 104 to 107 H. pylori CFU per gram of gastric mucus[5].
Upon infection, H. pylori uses urease and α-carbonic anhydrase to generate ammonia and HCO32- which mitigate the effects of low pH[6,7]. Moreover, thanks to its flagella and shape, H. pylori penetrate the mucus layer. H. pylori null mutant defective in production of flagella are unable to colonize gnotobiotic piglets[8]. Once established in the inner mucus layer, several outer membrane proteins, including BabA, SabA, AlpA, AlpB and HopZ can mediate bacterial adherence to gastric epithelial cells. Once attached, bacterial effector molecules, both secreted [vacuolating cytotoxin (VacA) and cytotoxin- associated gene A (CagA)] or attached [components of the type IV secretion system (CagL)], modulate gastric epithelial cell behaviour leading to loss of cell polarity, release of nutrients and chemokines [e.g., interleukin (IL)-8], and regulation of acid secretion via control of gastrin and H+/ K+ ATPase[9,10].
The infections are acquired during childhood; frequent clonal transmission of H. pylori between first degree relatives demonstrates intra-familial transmission of H. pylori in developed countries. In developing world, members of the same family can be infected with widely diverse strains, and multiple infections were common arguing for horizontal transmission of H. pylori infection[11]. After ingestion, there is a period of intense bacterial proliferation and gastric inflammation. Concomitant with the intense gastritis is hypochlorhydria. Fecal shedding of H. pylori is maximal during this period, facilitating transmission to new hosts. Ultimately, the inflammatory response is reduced to a low-level stable state, normal gastric pH is restored, and most of the infected person becomes asymptomatic[12]. This outcome persists for years or decades and appears to predominate in the population. Depending on H. pylori virulence factors, environmental factors and the host response to bacterial infection, H. pylori infection can be associated with several clinical complications such as gastritis, peptic ulcer disease, gastric cancer and mucosa-associated lymphoid tissue (MALT) lymphoma[13-15]. H. pylori eradication therapies have revolutionised the natural course of peptic ulcer disease[13]. Antibiotic treatment of H. pylori infection is relatively successful, with the organism being eradicated from around 80% of patients[16].
IMMUNE RESPONSE TO H. PYLORI INFECTION
Immune responses to H. pylori infection have been studied in twenty adult volunteers experimentally infected with H. pylori[17]. Gastric biopsies performed 2 wk after infection showed infiltration of lymphocytes and monocytes, along with significantly increased expression of IL-1, IL-8, and IL-6 in the gastric antrum[17]. Anti-H. pylori immunoglobulin (Ig)M and IgG responses were detected in the serum of infected individuals. In addition, 4 wk after infection, the numbers of gastric CD4+ and CD8+ T cells were increased compared to preinfection levels[18]. These data provide evidence that gastric and systemic immune responses develops within a short period of time after H. pylori infection.
Gastric mucosal biopsies from humans persistently infected with H. pylori reveal an increased infiltration of various types of leukocytes compared to biopsies from uninfected humans[19]. Lymphocytes (T and B cells), monocytes, eosinophils, macrophages, neutrophils, mast cells and dendritic cells are usually present[19,20]. B cells and CD4+ T cells together with dendritic cells (DC) sometimes organize into lymphoid follicles[21] reflecting ongoing antigen presentation and chronic immune responses. H. pylori-specific CD4+ T cells are detectable in the gastric mucosa and peripheral blood of infected-individuals but not uninfected humans[22]. Levels of cytokines [interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), IL-1, IL-6, IL-7, IL-8, IL-10, and IL-18] are increased in the stomach of H. pylori-infected humans compared to uninfected humans[23]. IL-4 has not been detected in the gastric mucosa of most H. pylori-infected individuals[24]. Therefore, it has been concluded that H. pylori infection leads to a T helper cell (Th)1-polarized response. H. pylori infection has also been associated with upregulation of IL-17A expression in the gastric mucosa[25]. IL-17A is the most widely studied member of the IL-17 family of cytokines (IL-17A-F), and is produced by Th17 CD4+ T cells as well as other subsets of immune cells[26]. Extracellular bacterial and fungal infections elicit strong IL-17A responses that stimulate stromal and epithelial cells to release pro-inflammatory cytokines and chemokines, e.g., TNF-α, IL-1β, IL-6, CXCL1, CXCL2, CCL2, CCL7, CCL20, which recruit neutrophils, macrophages and lymphocytes to the site of infection[27]. Furthermore, it has been described that H. pylori infection also leads to the generation of regulatory T cells (Treg)[28-30]. Depletion of Treg through injection of anti-CD25 antibodies to mice before H. pylori infection promoted gastritis and reduced bacterial load[31]. Very elegant studies originated from the group of PD Smith clearly showed that in children[30,32], H. pylori infection is associated with low Th17 and Th1 responses, high Treg response and reduced gastritis as compared with adults, suggesting that H. pylori specific Treg play key roles in bacterial persistence.
Associated with cellular responses, a humoral immune response is elicited in nearly all H. pylori-infected humans[33]. Serum IgA and IgG antibodies in chronically infected persons are directed toward many different H. pylori antigens[33]. A local antibody response directed toward H. pylori antigens is also detectable with chronic H. pylori infection. These subjects have remarkably higher frequencies of total IgA- and IgM-secreting cells than the noninfected subjects, while the frequencies of IgG-secreting cells were virtually the same in the different groups[34]. Notably, H. pylori infection induces autoantibodies reactive with gastric epithelial cells, which could drive gastritis[35]. These autoantibodies could be directly cytolytic to epithelial cells through activation of complement, inducing apoptosis or triggering an antibody-dependent cellular cytotoxicity reaction leading to the tissue destruction.
GASTRO-INTESTINAL TRACT IMMUNE DEFENCES
H. pylori colonizes the gastro-intestinal tract, thus there is a need to study the immune responses directed toward H. pylori in the context of the general functioning of the gastro-intestinal tract immune defences. In the following paragraph, we will briefly summarize our current understanding of the functioning of the mucosal immune responses.
The mucosal defences are multiple and might be physical, chemical and immune-mediated. The mucosal epithelium blocks invasion by pathogenic and commensal bacteria by forming multiple layers of physical (tight junctions), chemical nitric oxide and immune protection (local secretion of defensins, anti- and/or pro-inflammatory chemokines/cytokines and IgA/IgG/IgM transport). In addition, numerous bone marrow-derived cells belonging to the innate or adaptive immune systems colonized the intestinal mucosa to fight the invaders, but at steady state the same cells have to tolerate commensals.
IgA response
A major defensive mechanism that excludes commensals and pathogens from the mucosal surface involves IgA[36]. Mucosal IgA comprises antibodies that recognize antigens with high- and low-affinity binding modes. In general, high-affinity IgA neutralizes microbial toxins and invasive pathogens, whereas low-affinity IgA confines commensals in the intestinal lumen. High-affinity IgA is thought to emerge in Peyer’s Patches (PPs) and mesenteric lymph nodes (MLNs) from follicular B cells stimulated via T cell-dependent pathways, whereas low-affinity IgA likely emerges in PPs, MLNs and lamina propria from B cells stimulated via T cell-independent pathways[36]. IgA response is powerfully induced by the presence of commensal microbes in the intestine[37,38] and has been shown to promote the maintenance of appropriate bacterial communities in specific intestinal segments[39]. In contrast to the lungs, vagina and most of the gastrointestinal tract, the healthy mammalian stomach produces very low level of polymeric immunoglobulin receptor (pIgR)[40,41], the receptor mediating IgA transport into the gastrointestinal lumen. Studies in H. pylori-infected humans have shown that baseline pIgR expression by the gastric epithelium can be upregulated in response to gastric inflammation[42] due to increased local IFN-γ production[43]. However, despite significantly increased pIgR expression and IgA plasma cell infiltration in response to H. pylori infection[44] there is no concomitant increase in IgA secretion into the stomach; and it is non-secretory monomeric IgA which predominates in the stomach of H. pylori-infected individuals[45]. Hence, the IgA that is present in the gastric lumen would be unstable, susceptible to degradation by proteases. These observations suggest that, the stomach anti-H. pylori IgA responses do not play similar biological roles as compared with anti-commensal or anti-pathogen IgA response taking place in the intestine.
IgG response
In unmanipulated specific pathogen-free animals it has been showed that there was no specific serum IgG response detectable directed against commensal bacteria[46]. In pathogen-free mice, the systemic immune system appeared to remain ignorant of the commensal microbes. However, in human, a certain degree of systemic exposure to gut commensal bacteria and the associated priming of systemic immune response seems to be well tolerated, harmless and common in healthy humans since systemic antibody responses against live gut commensal bacteria and fungi can be detected[47]. Most of the H. pylori infected individuals develop systemic anti-H. pylori IgG responses[18]. Recently, Ben Suleiman et al[48] detected the expression of neonatal Fc receptor in gastric epithelial cells, this receptor was shown to transport IgG into gastric secretion. These results indicate that systemic anti-H. pylori IgG response might gain access to the gastric mucosa and exert some anti-bacterial and/or pro-inflammatory activities.
CD4+ T cell responses
Since H. pylori is an extra-cellular bacteria, anti-H. pylori specific CD8+ T cell responses are inadequate to protect the host against such pathogen. Hence, in this review we will only describe the priming of CD4+ T cell response. As discussed above for IgA response, CD4+ T cell responses are initiated within the PPs and MLNs. DC capture, process and present antigens to naive T cells in PPs and MLNs. In the stomach, DCs are penetrating the mucosa[49] to sample luminal antigens and migrate to the stomach lymph node[50].
At steady state, mucosal CD4+ T cells are tolerant to microbiota-derived antigens[51]. Remarkably, systemic CD4+ T cells are not tolerant to microbiota-derived antigens and conserved a naïve state to these antigens[52]. It has recently been suggested that antigen-specific intestinal IgA play a critical role in inhibiting the systemic CD4+ T-cell responses to commensal antigens by providing immune exclusion[51].
At mucosal surfaces, DCs maintain homeostasis by dampening inflammatory Th1 and Th17 cell responses[53]. Mucosal DCs are particularly skilled in eliciting these anti-inflammatory responses because they receive conditioning signals from intestinal epithelial cells (IECs)[54,55]. One of these signals is provided by thymic stromal lymphopoietin (TSLP), that shifts the Th1/Th2 balance toward Th2 polarization by attenuating DC production of IL-12 but not of IL-10[56]. In addition to TSLP, IECs release transforming growth factor (TGF)-β and retinoic acid, which stimulate the development of CD103+ DCs[53]. These DCs promote the formation of Treg cells via TGF-β and retinoic acid and suppress the development of inflammatory Th1 and Th17 cells[53].
In addition to initiating responses that create an overall tolerant state towards harmless intestinal antigens, mucosal DCs are also implicated in the generation of protective immune responses aimed at the clearance of enteric pathogens. A fundamental difference between the steady state and a state of infection may lie in the greater propensity of pathogens to invade and penetrate beneath the epithelial-cell layer. Invasion of IECs would allow for the activation of cytosolic pattern-recognition receptors, TLRs and both quantitative and qualitative changes in the secretion of pro-inflammatory cytokines and chemokines. Consistent with this, IECs produce CXC-chemokine ligand 8 (CXCL8) when infected with strains of Salmonella spp. that are both invasive and flagellated[57]. CXCL8 may serve to attract neutrophils to the site of infection, furthering the inflammatory milieu. As a result, the rate of blood-borne DC precursors migrating into the tissues and becoming DCs will increase. These cells will not have been subjected to IECs conditioning and can be directly activated by a combination of pathogens that have breached the epithelial-cell barrier and the pro-inflammatory cytokine milieu. Experimental data support this scenario; human monocyte-derived DCs conditioned with IEC supernatants are impaired in their ability to secrete IL-12 and drive Th1-cell responses following exposure to pathogenic Salmonella spp[56] but can drive Th1-cell responses if they encounter bacteria before conditioning by IEC-derived factors. One other possible route for the generation of protective immunity to pathogens may be the uptake of pathogenic species by DCs that are normally resident in the MLNs. In this respect, CD103- MLN DCs have been shown to produce higher levels of pro-inflammatory cytokines than their intestinal-derived CD103+ counterparts and drive IFNγ and IL-17 production by CD4+ T cells[53].
Collectively, since H. pylori is mostly a non-invasive bacteria living within the stomach mucosa, these observations suggest that, the CD4+ T cells responses directed against H. pylori, initiated within PPs, MLNs and stomach draining lymph node, might be naturally more tolerogenic than pro-inflammatory. This assumption is corroborate by the recent demonstration that in children, H. pylori infection is associated with low Th17 and Th1 responses and high Treg response[32]. However, the detection of H. pylori specific Th17/Th1 in chronically infected individuals[24,25] shows that the initial tolerogenic response is progressively lost, showing that with time the mucosal immune system identified H. pylori as a pathogen.
Intra-epithelial lymphocytes, innate immune cells and others
Intra-epithelial cells[58], innate immune cells[59], natural killer cells[60], neutrophils[61], mast cells[62], eosinophils[63], macrophages[64], monocytes[64], suppressive myeloid cells[65] are playing roles in the functioning of the mucosal immune system, however due to space limitation we will not discuss their roles in the context of H. pylori infection.
STOMACH MICROBIOTA
It was previously admitted that the stomach was a sterile organ and that pH values < 4, peristalsis and high bile concentration were able to sterilize the stomach, but in the past 30 years with the discovery of H. pylori it is now known that the stomach supports a bacterial community with hundreds of phylotypes[66-68] Although, the stomach, along with the esophagus and the duodenum, are the least colonized regions of the gastro-intestinal (GI) tract, in contrast to the high bacterial counts (1010 to 1012 CFU/g) observed in the colon. While it has been postulated that the indigenous stomach microbiota might be a reflection of transient bacteria from the mouth and esophagus, three separate studies demonstrated that in spite of high inter-subject variability, the gastric microbiota were distinguishable from microbiota found in the mouth, nose, and distal GI tract[69]. The most abundant phyla in H. pylori positive subjects are Proteobacteria, Firmicutes and Actinobacteria. In the absence of H. pylori, the most abundant phyla are Firmicutes, Bacteroidetes and Actinobacteria[69].
In the gastro-intestinal tract, the microbiota has a major impact on the functioning of the mucosal immune system and vice versa. Germ-free mice have small size of PPs, decrease number of lamina propria IgA secreting-plasmocytes, low levels of serum immunoglobulin and demonstrate no Th17/Th1 in the intestine[70]. The composition of the intestinal flora modulates the functioning of the immune system, for instance, the presence of Segmented filamentous bacteria (SFB) in the microbiota is associated with the development of Th17 in the intestinal lamina propria[71]. The presence of some Clostridia strains within the human intestinal microbiota has been recently associated with the development of intestinal Treg[72]. In addition, some commensal bacteria and microbiota-derived metabolites like short-chain fatty acids have been shown to inhibit inflammatory reactions at intestinal levels and promote pathogen clearance[59,73,74]. Inversely, defects in antibody response lead to a modification of the bacterial composition of the intestinal flora[39]. Collectively, these observations suggest that the colonization of the stomach mucosa by H. pylori and/or the associated microbiota might also impact the functioning of the immune system of the host and vice versa.
TOLEROGENIC ACTIVITIES OF H. PYLORI
Studies indicate that H. pylori-derived factors are capable to inhibit T-cell proliferation. Using normal T cells and Jurkat cells, a human T-cell line, it was demonstrated that VacA interfered with calcium-signaling events inside the cell and prevented activation of the calcium-dependent phosphatase calcineurin[75,76]. The subsequent dephosphorylation of nuclear factor of activated T cells (NFAT), a transcription factor that regulates immune responses, was suppressed resulting in inhibition of IL-2 expression and proliferation of T cells. Similar anti-proliferative effect on T cells was reported for the γ-glutamyl transpeptidase (γ-GGT), this immunosuppressive factor inhibits T-cell proliferation by induction of a cell cycle arrest in the G(1) phase[77]. In addition to VacA and γ-GGT, H. pylori arginase can impair T-cell function during infection. Using Jurkat T cells and human normal lymphocytes, it was found that a wild type H. pylori strain, but not an arginase mutant strain, inhibited T-cell proliferation, depleted L-arginine, and reduced the expression of the CD3 chain of the T-cell receptor[78]. Most (80%-90%) H. pylori strains display Lewis blood-group antigens on their LPS, and these are similar to the Lewis blood-group antigens that are expressed on the mucosal surface of the human stomach[79]. Lewis positive H. pylori variants are able to bind to the C-type lectin DC-SIGN and present on gastric DCs, and demonstrate that this interaction blocks Th1 development[80].
In addition to suppress T cell activation, H. pylori has been demonstrated to decrease the functioning of the innate immune system. For instance, efficient phagocytosis and killing of H. pylori is prevented by the presence of the cag pathogenicity island[81,82] and H. pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity[83]. Importantly, at the opposite to the LPS and flagellins of others gram-negative bacteria, the LPS and flagellins of H. pylori do not adequately activate the antigen presenting cells via the Toll-like receptors[84,85].
Collectively, H. pylori counteract innate and T cell responses and clearly exhibited tolerogenic activities on the immune system. It can be suggested that these tolerogenic activities participate to the H. pylori persistence within the stomach mucosa.
VACCINE-INDUCED PROTECTIVE IMMUNE RESPONSES
H. pylori infection is the main cause of gastritis, peptic ulcers, and gastric adenocarcinoma. It is believed that H. pylori contributes to gastric cancer development by direct action of its virulence factors and indirectly by initiation and maintenance of a chronic inflammation in the gastric mucosa[86]. Hence, gastroenterologists use a combination of anti-secretory and antimicrobial agents to eradicate H. pylori[16]. Similar to other antimicrobial treatments, the therapy may select resistant H. pylori strains[16]. Therefore, alternative therapies to eradicate H. pylori infection have been evaluated like the development of a vaccine against H. pylori.
In the seminal work reported in 1990 by Lee et al[87] demonstrated the feasibility to study different aspects of the pathology and the immune response induced by Helicobacter species in mice. These investigators using germ-free mice and H. felis, a bacteria that naturally infects cats and dogs, achieved successful long-term colonization and associated gastritis in these mice. This model became very popular and a large number of immunization studies were performed in H. felis infected mice. This was made possible by the fact that vaccine candidate antigens are shared between H. felis and H. pylori species (i.e., urease and heat shock proteins). Thereafter, H. pylori strains have been adapted to the mouse stomach and this experimental model reproduces several aspects of the human infection[88-90]. Successful colonization with H. pylori has been reported in rats, guinea pigs, Mongolian gerbils, Gnotobiotic pigs, cats and Beagle dogs[90]. H. pylori naturally infects some species of nonhuman primates, with pathological changes in the stomach resulting from H. pylori infection being very similar to those observed in humans[91].
Numerous studies in animals suggested that T cells, mast cells and neutrophils are of prime importance for protection, while B cells (antibodies) are dispensable for protection[61,62,92,93]. However some studies suggested that antibodies can also participate to the vaccine-induced H. pylori clearance in some circumstances[94-98]. Indeed, vaccination-induced protection against H. pylori in mice requires major histocompatibility complex class II-restricted CD4+ T cells[90,92], Th-1, Th-2 and/or Th-17 CD4+ T cell responses and the α4β7 integrin-mediated homing process[99] have been implicated in protection[100-103]. Recently, the production of IL-17, by Th17 cells, has been clearly identified as a key player in the vaccine-induced H. pylori clearance. IL-17 has also been linked to neutrophil recruitment and activation through the induction of granulocyte-stimulating factor and IL-8[104] and to resistance against extracellular microbial infections[105], leading to the conclusion that IL-17 production by H. pylori specific Th17 cells can mediate the vaccine-induced H. pylori clearance (Figure 1).
Figure 1.
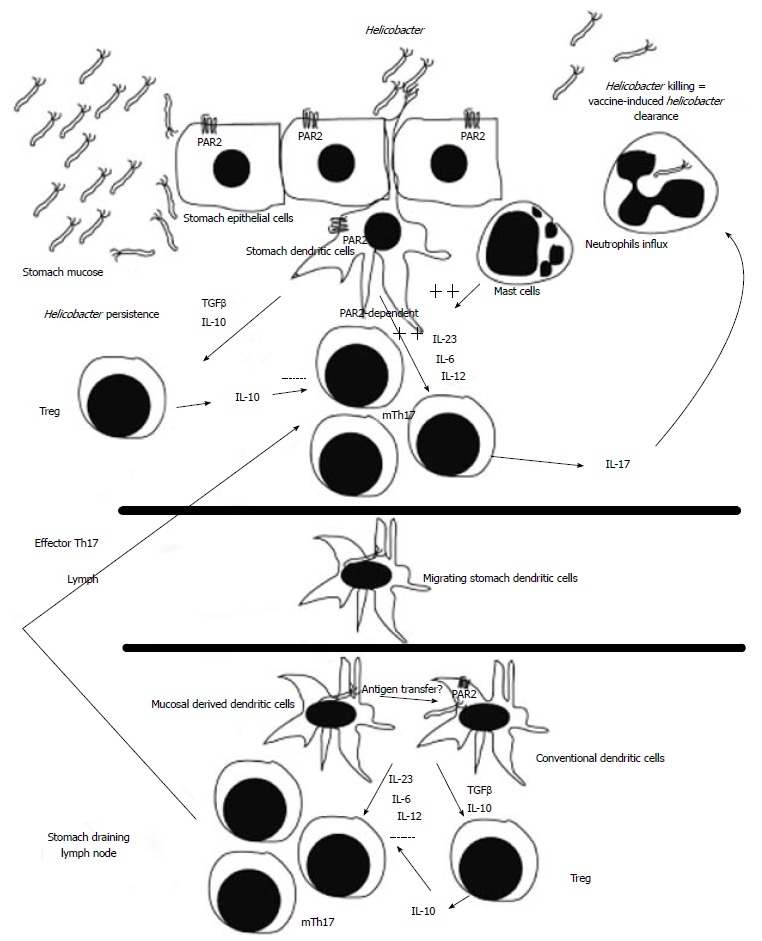
Schematic representation of the vaccine-induced Helicobacter pylori clearance. During Helicobacter infection of vaccinated hosts, memory T helper (Th)17 cells (mTh17) are primed by protease-activated receptor (PAR)2-dependent dendritic cell (DC)[126] directly in the stomach and/or in the stomach draining lymph nodes (conventional DCs). Effector memory Th17 cells originated from the stomach and/or from the stomach draining lymph nodes will produce high levels of interleukin (IL)-17 leading to recruitment of neutrophils and to Helicobacter clearance. In naïve hosts, DCs mainly prime regulatory T cells (Treg), leading to Helicobacter persistence.
Collectively, it was clearly demonstrated that H. pylori infections could be substantially prevented, reduced or even eliminated by prophylactic and therapeutic mucosal and systemic vaccinations[106-110]. This result is of great interest not only for the development of H. pylori vaccine but also for vaccine strategy aimed at clearing commensal bacteria with genotoxic and mutagenic activities[111].
CONCLUSION
H. pylori can be considered as a commensal bacteria belonging to stomach microbiota. Indeed, H. pylori promote the generation of H. pylori specific Treg. The tolerogenic environment created by H. pylori might explain that H. pylori seropositivity was inversely related to recent wheezing, allergic rhinitis, dermatitis, eczema, asthma or rash[112]. Very elegant pre-clinical studies conducted by the group of A Müller recently gave support to this assumption by showing that H. pylori infection during the neonatal period promote the development of Treg responses that protect adult mice from asthma[113]. Hence, H. pylori, like other commensal bacteria such as SFB[71], Calibacterium prausnitzi[74], Lactobacillus reuteri[59], Lactobacillus Acidophilus[59], and Clostridia[72] profoundly impact the functioning of the immune system of the colonized host.
Although H. pylori infection can be beneficial for the host, when present in the stomach, H. pylori reduce the overall diversity of the gastric microbiota[114] and promote gastritis. The modification of the stomach microbiota might, independently or not of the presence of H. pylori, modulate the susceptibility of the host to immune-mediated diseases. The H. pylori-induced gastritis is most probably cause by the type IV secretion apparatus-dependent introduction of muropeptides into epithelial cells, that promote Nod1-dependent induction of a proinflammatory program[115]. In addition H. pylori promote gastric inflammation through the production of a neutrophil activating protein[116]. In spite of the natural tolerogenic environment provided by the stomach mucosa and the tolerogenic activities of H. pylori, these pro-inflammatory signals initiating systemic and local pro-inflammatory Th1/Th17 responses[22-24].
Since H. pylori possess pro-carcinogenic activities via maintenance of a chronic inflammation in the gastric mucosa and by direct action of its virulence factors (vacA and cagA), H. pylori cannot be considered as symbiotic bacteria but rather as part of the pathobiont[117]. Hence, H. pylori has to be eliminated when individuals are prone to develop duodenal and stomach ulcers[118,119] to prevent further major diseases development like MALT lymphoma and stomach adenocarcinoma. Although, the design of a vaccine directed H. pylori is challenging since it has to overcome the natural tolerogenic environment provided by the stomach mucosa and the tolerogenic activities of H. pylori, its development will bring health benefits for individuals infected with antibiotic resistant H. pylori strains and population of underdeveloped countries[120-125].
Footnotes
Supported by the Swiss National Foundation grants 310030_141145, to Velin D
P- Reviewers: Cardaropoli S, Cong WM S- Editor: Gou SX L- Editor: A E- Editor: Liu XM
References
- 1.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, Werling HO, Josenhans C, Suerbaum S. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc Natl Acad Sci USA. 2004;101:5024–5029. doi: 10.1073/pnas.0308386101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hazell SL, Lee A, Brady L, Hennessy W. Campylobacter pyloridis and gastritis: association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J Infect Dis. 1986;153:658–663. doi: 10.1093/infdis/153.4.658. [DOI] [PubMed] [Google Scholar]
- 4.Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and interstitial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J Infect Dis. 2003;187:1165–1177. doi: 10.1086/368133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atherton JC, Tham KT, Peek RM, Cover TL, Blaser MJ. Density of Helicobacter pylori infection in vivo as assessed by quantitative culture and histology. J Infect Dis. 1996;174:552–556. doi: 10.1093/infdis/174.3.552. [DOI] [PubMed] [Google Scholar]
- 6.Bauerfeind P, Garner R, Dunn BE, Mobley HL. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut. 1997;40:25–30. doi: 10.1136/gut.40.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. The HP0165-HP0166 two-component system (ArsRS) regulates acid-induced expression of HP1186 alpha-carbonic anhydrase in Helicobacter pylori by activating the pH-dependent promoter. J Bacteriol. 2007;189:2426–2434. doi: 10.1128/JB.01492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eaton KA, Suerbaum S, Josenhans C, Krakowka S. Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. Infect Immun. 1996;64:2445–2448. doi: 10.1128/iai.64.7.2445-2448.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiedemann T, Hofbaur S, Tegtmeyer N, Huber S, Sewald N, Wessler S, Backert S, Rieder G. Helicobacter pylori CagL dependent induction of gastrin expression via a novel αvβ5-integrin-integrin linked kinase signalling complex. Gut. 2012;61:986–996. doi: 10.1136/gutjnl-2011-300525. [DOI] [PubMed] [Google Scholar]
- 10.Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology. 2010;139:239–248. doi: 10.1053/j.gastro.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarz S, Morelli G, Kusecek B, Manica A, Balloux F, Owen RJ, Graham DY, van der Merwe S, Achtman M, Suerbaum S. Horizontal versus familial transmission of Helicobacter pylori. PLoS Pathog. 2008;4:e1000180. doi: 10.1371/journal.ppat.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blaser MJ, Parsonnet J. Parasitism by the “slow” bacterium Helicobacter pylori leads to altered gastric homeostasis and neoplasia. J Clin Invest. 1994;94:4–8. doi: 10.1172/JCI117336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ford A, Delaney B, Forman D, Moayyedi P. Eradication therapy for peptic ulcer disease in Helicobacter pylori positive patients. Cochrane Database Syst Rev. 2004;(4):CD003840. doi: 10.1002/14651858.CD003840.pub2. [DOI] [PubMed] [Google Scholar]
- 14.Fukase K, Kato M, Kikuchi S, Inoue K, Uemura N, Okamoto S, Terao S, Amagai K, Hayashi S, Asaka M. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008;372:392–397. doi: 10.1016/S0140-6736(08)61159-9. [DOI] [PubMed] [Google Scholar]
- 15.Farinha P, Gascoyne RD. Helicobacter pylori and MALT lymphoma. Gastroenterology. 2005;128:1579–1605. doi: 10.1053/j.gastro.2005.03.083. [DOI] [PubMed] [Google Scholar]
- 16.Wolle K, Malfertheiner P. Treatment of Helicobacter pylori. Best Pract Res Clin Gastroenterol. 2007;21:315–324. doi: 10.1016/j.bpg.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Graham DY, Opekun AR, Osato MS, El-Zimaity HM, Lee CK, Yamaoka Y, Qureshi WA, Cadoz M, Monath TP. Challenge model for Helicobacter pylori infection in human volunteers. Gut. 2004;53:1235–1243. doi: 10.1136/gut.2003.037499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nurgalieva ZZ, Conner ME, Opekun AR, Zheng CQ, Elliott SN, Ernst PB, Osato M, Estes MK, Graham DY. B-cell and T-cell immune responses to experimental Helicobacter pylori infection in humans. Infect Immun. 2005;73:2999–3006. doi: 10.1128/IAI.73.5.2999-3006.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;20:1161–1181. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki T, Kato K, Ohara S, Noguchi K, Sekine H, Nagura H, Shimosegawa T. Localization of antigen-presenting cells in Helicobacter pylori-infected gastric mucosa. Pathol Int. 2002;52:265–271. doi: 10.1046/j.1440-1827.2002.01347.x. [DOI] [PubMed] [Google Scholar]
- 21.Terrés AM, Pajares JM. An increased number of follicles containing activated CD69+ helper T cells and proliferating CD71+ B cells are found in H. pylori-infected gastric mucosa. Am J Gastroenterol. 1998;93:579–583. doi: 10.1111/j.1572-0241.1998.168_b.x. [DOI] [PubMed] [Google Scholar]
- 22.Di Tommaso A, Xiang Z, Bugnoli M, Pileri P, Figura N, Bayeli PF, Rappuoli R, Abrignani S, De Magistris MT. Helicobacter pylori-specific CD4+ T-cell clones from peripheral blood and gastric biopsies. Infect Immun. 1995;63:1102–1106. doi: 10.1128/iai.63.3.1102-1106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindholm C, Quiding-Järbrink M, Lönroth H, Hamlet A, Svennerholm AM. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–5971. doi: 10.1128/iai.66.12.5964-5971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D'Elios MM, Manghetti M, De Carli M, Costa F, Baldari CT, Burroni D, Telford JL, Romagnani S, Del Prete G. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J Immunol. 1997;158:962–967. [PubMed] [Google Scholar]
- 25.Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- 26.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 27.Cooper AM. IL-17 and anti-bacterial immunity: protection versus tissue damage. Eur J Immunol. 2009;39:649–652. doi: 10.1002/eji.200839090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun. 2003;71:1755–1762. doi: 10.1128/IAI.71.4.1755-1762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raghavan S, Fredriksson M, Svennerholm AM, Holmgren J, Suri-Payer E. Absence of CD4+CD25+ regulatory T cells is associated with a loss of regulation leading to increased pathology in Helicobacter pylori-infected mice. Clin Exp Immunol. 2003;132:393–400. doi: 10.1046/j.1365-2249.2003.02177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris PR, Wright SW, Serrano C, Riera F, Duarte I, Torres J, Peña A, Rollán A, Viviani P, Guiraldes E, et al. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology. 2008;134:491–499. doi: 10.1053/j.gastro.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 31.Rad R, Brenner L, Bauer S, Schwendy S, Layland L, da Costa CP, Reindl W, Dossumbekova A, Friedrich M, Saur D, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006;131:525–537. doi: 10.1053/j.gastro.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Serrano C, Wright SW, Bimczok D, Shaffer CL, Cover TL, Venegas A, Salazar MG, Smythies LE, Harris PR, Smith PD. Downregulated Th17 responses are associated with reduced gastritis in Helicobacter pylori-infected children. Mucosal Immunol. 2013;6:950–959. doi: 10.1038/mi.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez-Perez GI, Dworkin BM, Chodos JE, Blaser MJ. Campylobacter pylori antibodies in humans. Ann Intern Med. 1988;109:11–17. doi: 10.7326/0003-4819-109-1-11. [DOI] [PubMed] [Google Scholar]
- 34.Mattsson A, Quiding-Järbrink M, Lönroth H, Hamlet A, Ahlstedt I, Svennerholm A. Antibody-secreting cells in the stomachs of symptomatic and asymptomatic Helicobacter pylori-infected subjects. Infect Immun. 1998;66:2705–2712. doi: 10.1128/iai.66.6.2705-2712.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Claeys D, Faller G, Appelmelk BJ, Negrini R, Kirchner T. The gastric H+,K+-ATPase is a major autoantigen in chronic Helicobacter pylori gastritis with body mucosa atrophy. Gastroenterology. 1998;115:340–347. doi: 10.1016/s0016-5085(98)70200-8. [DOI] [PubMed] [Google Scholar]
- 36.Cerutti A, Chen K, Chorny A. Immunoglobulin responses at the mucosal interface. Annu Rev Immunol. 2011;29:273–293. doi: 10.1146/annurev-immunol-031210-101317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benveniste J, Lespinats G, Salomon J. Serum and secretory IgA in axenic and holoxenic mice. J Immunol. 1971;107:1656–1662. [PubMed] [Google Scholar]
- 38.Benveniste J, Lespinats G, Adam C, Salomon JC. Immunoglobulins in intact, immunized, and contaminated axenic mice: study of serum IgA. J Immunol. 1971;107:1647–1655. [PubMed] [Google Scholar]
- 39.Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, Fagarasan S. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci USA. 2004;101:1981–1986. doi: 10.1073/pnas.0307317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaneko T, Ota H, Hayama M, Akamatsu T, Katsuyama T. Helicobacter pylori infection produces expression of a secretory component in gastric mucous cells. Virchows Arch. 2000;437:514–520. doi: 10.1007/s004280000285. [DOI] [PubMed] [Google Scholar]
- 41.Isaacson P. Immunoperoxidase study of the secretory immunoglobulin system and lysozyme in normal and diseased gastric mucosa. Gut. 1982;23:578–588. doi: 10.1136/gut.23.7.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahlstedt I, Lindholm C, Lönroth H, Hamlet A, Svennerholm AM, Quiding-Järbrink M. Role of local cytokines in increased gastric expression of the secretory component in Helicobacter pylori infection. Infect Immun. 1999;67:4921–4925. doi: 10.1128/iai.67.9.4921-4925.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer F, Wilson KT, James SP. Modulation of innate cytokine responses by products of Helicobacter pylori. Infect Immun. 2000;68:6265–6272. doi: 10.1128/iai.68.11.6265-6272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quiding-Järbrink M, Sundström P, Lundgren A, Hansson M, Bäckström M, Johansson C, Enarsson K, Hermansson M, Johnsson E, Svennerholm AM. Decreased IgA antibody production in the stomach of gastric adenocarcinoma patients. Clin Immunol. 2009;131:463–471. doi: 10.1016/j.clim.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 45.Birkholz S, Schneider T, Knipp U, Stallmach A, Zeitz M. Decreased Helicobacter pylori-specific gastric secretory IgA antibodies in infected patients. Digestion. 1998;59:638–645. doi: 10.1159/000007568. [DOI] [PubMed] [Google Scholar]
- 46.Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–2226. doi: 10.1126/science.288.5474.2222. [DOI] [PubMed] [Google Scholar]
- 47.Zimmermann K, Haas A, Oxenius A. Systemic antibody responses to gut microbes in health and disease. Gut Microbes. 2012;3:42–47. doi: 10.4161/gmic.19344. [DOI] [PubMed] [Google Scholar]
- 48.Ben Suleiman Y, Yoshida M, Nishiumi S, Tanaka H, Mimura T, Nobutani K, Yamamoto K, Takenaka M, Aoganghua A, Miki I, et al. Neonatal Fc receptor for IgG (FcRn) expressed in the gastric epithelium regulates bacterial infection in mice. Mucosal Immunol. 2012;5:87–98. doi: 10.1038/mi.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–1054. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scheinecker C, McHugh R, Shevach EM, Germain RN. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J Exp Med. 2002;196:1079–1090. doi: 10.1084/jem.20020991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng T, Elson CO. Adaptive immunity in the host-microbiota dialog. Mucosal Immunol. 2011;4:15–21. doi: 10.1038/mi.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konrad A, Cong Y, Duck W, Borlaza R, Elson CO. Tight mucosal compartmentation of the murine immune response to antigens of the enteric microbiota. Gastroenterology. 2006;130:2050–2059. doi: 10.1053/j.gastro.2006.02.055. [DOI] [PubMed] [Google Scholar]
- 53.Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8:435–446. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rescigno M, Lopatin U, Chieppa M. Interactions among dendritic cells, macrophages, and epithelial cells in the gut: implications for immune tolerance. Curr Opin Immunol. 2008;20:669–675. doi: 10.1016/j.coi.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 55.Rescigno M, Di Sabatino A. Dendritic cells in intestinal homeostasis and disease. J Clin Invest. 2009;119:2441–2450. doi: 10.1172/JCI39134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rimoldi M, Chieppa M, Salucci V, Avogadri F, Sonzogni A, Sampietro GM, Nespoli A, Viale G, Allavena P, Rescigno M. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol. 2005;6:507–514. doi: 10.1038/ni1192. [DOI] [PubMed] [Google Scholar]
- 57.Rimoldi M, Chieppa M, Larghi P, Vulcano M, Allavena P, Rescigno M. Monocyte-derived dendritic cells activated by bacteria or by bacteria-stimulated epithelial cells are functionally different. Blood. 2005;106:2818–2826. doi: 10.1182/blood-2004-11-4321. [DOI] [PubMed] [Google Scholar]
- 58.Trejdosiewicz LK, Calabrese A, Smart CJ, Oakes DJ, Howdle PD, Crabtree JE, Losowsky MS, Lancaster F, Boylston AW. Gamma delta T cell receptor-positive cells of the human gastrointestinal mucosa: occurrence and V region gene expression in Heliobacter pylori-associated gastritis, coeliac disease and inflammatory bowel disease. Clin Exp Immunol. 1991;84:440–444. [PMC free article] [PubMed] [Google Scholar]
- 59.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–385. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 60.Lindgren Å, Yun CH, Sjöling Å, Berggren C, Sun JB, Jonsson E, Holmgren J, Svennerholm AM, Lundin SB. Impaired IFN-γ production after stimulation with bacterial components by natural killer cells from gastric cancer patients. Exp Cell Res. 2011;317:849–858. doi: 10.1016/j.yexcr.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 61.Ismail HF, Fick P, Zhang J, Lynch RG, Berg DJ. Depletion of neutrophils in IL-10(-/-) mice delays clearance of gastric Helicobacter infection and decreases the Th1 immune response to Helicobacter. J Immunol. 2003;170:3782–3789. doi: 10.4049/jimmunol.170.7.3782. [DOI] [PubMed] [Google Scholar]
- 62.Velin D, Bachmann D, Bouzourene H, Michetti P. Mast cells are critical mediators of vaccine-induced Helicobacter clearance in the mouse model. Gastroenterology. 2005;129:142–155. doi: 10.1053/j.gastro.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 63.Garhart CA, Nedrud JG, Heinzel FP, Sigmund NE, Czinn SJ. Vaccine-induced protection against Helicobacter pylori in mice lacking both antibodies and interleukin-4. Infect Immun. 2003;71:3628–3633. doi: 10.1128/IAI.71.6.3628-3633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fiocca R, Luinetti O, Villani L, Chiaravalli AM, Capella C, Solcia E. Epithelial cytotoxicity, immune responses, and inflammatory components of Helicobacter pylori gastritis. Scand J Gastroenterol Suppl. 1994;205:11–21. [PubMed] [Google Scholar]
- 65.Ericksen RE, Rose S, Westphalen CB, Shibata W, Muthupalani S, Tailor Y, Friedman RA, Han W, Fox JG, Ferrante AW, et al. Obesity accelerates Helicobacter felis-induced gastric carcinogenesis by enhancing immature myeloid cell trafficking and TH17 response. Gut. 2014;63:385–394. doi: 10.1136/gutjnl-2013-305092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li XX, Wong GL, To KF, Wong VW, Lai LH, Chow DK, Lau JY, Sung JJ, Ding C. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS One. 2009;4:e7985. doi: 10.1371/journal.pone.0007985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sheh A, Fox JG. The role of the gastrointestinal microbiome in Helicobacter pylori pathogenesis. Gut Microbes. 2013;4:505–531. doi: 10.4161/gmic.26205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 73.Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amedei A, D’Elios MM, Telford JL, Baldari CT. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med. 2003;198:1887–1897. doi: 10.1084/jem.20030621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099–1102. doi: 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 77.Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, Bauer S, Brenner L, Schmid RM, Gerhard M. Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology. 2007;132:1820–1833. doi: 10.1053/j.gastro.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 78.Zabaleta J, McGee DJ, Zea AH, Hernández CP, Rodriguez PC, Sierra RA, Correa P, Ochoa AC. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR zeta-chain (CD3zeta) J Immunol. 2004;173:586–593. doi: 10.4049/jimmunol.173.1.586. [DOI] [PubMed] [Google Scholar]
- 79.Simoons-Smit IM, Appelmelk BJ, Verboom T, Negrini R, Penner JL, Aspinall GO, Moran AP, Fei SF, Shi BS, Rudnica W, et al. Typing of Helicobacter pylori with monoclonal antibodies against Lewis antigens in lipopolysaccharide. J Clin Microbiol. 1996;34:2196–2200. doi: 10.1128/jcm.34.9.2196-2200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth HP, Kapsenberg ML, Vandenbroucke-Grauls CM, van Kooyk Y, Appelmelk BJ. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J Exp Med. 2004;200:979–990. doi: 10.1084/jem.20041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allen LA, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med. 2000;191:115–128. doi: 10.1084/jem.191.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ramarao N, Gray-Owen SD, Backert S, Meyer TF. Helicobacter pylori inhibits phagocytosis by professional phagocytes involving type IV secretion components. Mol Microbiol. 2000;37:1389–1404. doi: 10.1046/j.1365-2958.2000.02089.x. [DOI] [PubMed] [Google Scholar]
- 83.Ramarao N, Gray-Owen SD, Meyer TF. Helicobacter pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity. Mol Microbiol. 2000;38:103–113. doi: 10.1046/j.1365-2958.2000.02114.x. [DOI] [PubMed] [Google Scholar]
- 84.Gewirtz AT, Yu Y, Krishna US, Israel DA, Lyons SL, Peek RM. Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J Infect Dis. 2004;189:1914–1920. doi: 10.1086/386289. [DOI] [PubMed] [Google Scholar]
- 85.Suda Y, Kim YM, Ogawa T, Yasui N, Hasegawa Y, Kashihara W, Shimoyama T, Aoyama K, Nagata K, Tamura T, et al. Chemical structure and biological activity of a lipid A component from Helicobacter pylori strain 206. J Endotoxin Res. 2001;7:95–104. [PubMed] [Google Scholar]
- 86.Correa P. Human gastric carcinogenesis: a multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–6740. [PubMed] [Google Scholar]
- 87.Lee A, Fox JG, Otto G, Murphy J. A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology. 1990;99:1315–1323. doi: 10.1016/0016-5085(90)91156-z. [DOI] [PubMed] [Google Scholar]
- 88.Marchetti M, Aricò B, Burroni D, Figura N, Rappuoli R, Ghiara P. Development of a mouse model of Helicobacter pylori infection that mimics human disease. Science. 1995;267:1655–1658. doi: 10.1126/science.7886456. [DOI] [PubMed] [Google Scholar]
- 89.Lee A, O’Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology. 1997;112:1386–1397. doi: 10.1016/s0016-5085(97)70155-0. [DOI] [PubMed] [Google Scholar]
- 90.Del Giudice G, Covacci A, Telford JL, Montecucco C, Rappuoli R. The design of vaccines against Helicobacter pylori and their development. Annu Rev Immunol. 2001;19:523–563. doi: 10.1146/annurev.immunol.19.1.523. [DOI] [PubMed] [Google Scholar]
- 91.Dubois A, Fiala N, Heman-Ackah LM, Drazek ES, Tarnawski A, Fishbein WN, Perez-Perez GI, Blaser MJ. Natural gastric infection with Helicobacter pylori in monkeys: a model for spiral bacteria infection in humans. Gastroenterology. 1994;106:1405–1417. doi: 10.1016/0016-5085(94)90392-1. [DOI] [PubMed] [Google Scholar]
- 92.Ermak TH, Giannasca PJ, Nichols R, Myers GA, Nedrud J, Weltzin R, Lee CK, Kleanthous H, Monath TP. Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J Exp Med. 1998;188:2277–2288. doi: 10.1084/jem.188.12.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Velin D, Bachmann D, Bouzourene H, Michetti P. Reduction of Helicobacter infection in IL-10-/- mice is dependent on CD4+ T cells but not on mast cells. Helicobacter. 2008;13:361–369. doi: 10.1111/j.1523-5378.2008.00614.x. [DOI] [PubMed] [Google Scholar]
- 94.Lee CK, Weltzin R, Thomas WD, Kleanthous H, Ermak TH, Soman G, Hill JE, Ackerman SK, Monath TP. Oral immunization with recombinant Helicobacter pylori urease induces secretory IgA antibodies and protects mice from challenge with Helicobacter felis. J Infect Dis. 1995;172:161–172. doi: 10.1093/infdis/172.1.161. [DOI] [PubMed] [Google Scholar]
- 95.Gorrell RJ, Wijburg OL, Pedersen JS, Walduck AK, Kwok T, Strugnell RA, Robins-Browne RM. Contribution of secretory antibodies to intestinal mucosal immunity against Helicobacter pylori. Infect Immun. 2013;81:3880–3893. doi: 10.1128/IAI.01424-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kleanthous H, Lee CK, Monath TP. Vaccine development against infection with Helicobacter pylori. Br Med Bull. 1998;54:229–241. doi: 10.1093/oxfordjournals.bmb.a011673. [DOI] [PubMed] [Google Scholar]
- 97.Thomas JE, Austin S, Dale A, McClean P, Harding M, Coward WA, Weaver LT. Protection by human milk IgA against Helicobacter pylori infection in infancy. Lancet. 1993;342:121. doi: 10.1016/0140-6736(93)91327-i. [DOI] [PubMed] [Google Scholar]
- 98.Corthésy-Theulaz I, Corthésy B, Bachmann D, Velin D, Kraehenbuhl JP. Passive immunity in Helicobacter-challenged neonatal mice conferred by immunized dams lasts until weaning. Infect Immun. 2003;71:2226–2229. doi: 10.1128/IAI.71.4.2226-2229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Michetti M, Kelly CP, Kraehenbuhl JP, Bouzourene H, Michetti P. Gastric mucosal alpha(4)beta(7)-integrin-positive CD4 T lymphocytes and immune protection against helicobacter infection in mice. Gastroenterology. 2000;119:109–118. doi: 10.1053/gast.2000.8548. [DOI] [PubMed] [Google Scholar]
- 100.Aebischer T, Laforsch S, Hurwitz R, Brombacher F, Meyer TF. Immunity against Helicobacter pylori: significance of interleukin-4 receptor alpha chain status and gender of infected mice. Infect Immun. 2001;69:556–558. doi: 10.1128/IAI.69.1.556-558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sawai N, Kita M, Kodama T, Tanahashi T, Yamaoka Y, Tagawa Y, Iwakura Y, Imanishi J. Role of gamma interferon in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Infect Immun. 1999;67:279–285. doi: 10.1128/iai.67.1.279-285.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.DeLyria ES, Redline RW, Blanchard TG. Vaccination of mice against H pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology. 2009;136:247–256. doi: 10.1053/j.gastro.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Velin D, Favre L, Bernasconi E, Bachmann D, Pythoud C, Saiji E, Bouzourene H, Michetti P. Interleukin-17 is a critical mediator of vaccine-induced reduction of Helicobacter infection in the mouse model. Gastroenterology. 2009;136:2237–2246.e1. doi: 10.1053/j.gastro.2009.02.077. [DOI] [PubMed] [Google Scholar]
- 104.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 105.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Czinn SJ, Cai A, Nedrud JG. Protection of germ-free mice from infection by Helicobacter felis after active oral or passive IgA immunization. Vaccine. 1993;11:637–642. doi: 10.1016/0264-410x(93)90309-l. [DOI] [PubMed] [Google Scholar]
- 107.Doidge C, Crust I, Lee A, Buck F, Hazell S, Manne U. Therapeutic immunisation against Helicobacter infection. Lancet. 1994;343:914–915. doi: 10.1016/s0140-6736(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 108.Corthésy-Theulaz I, Porta N, Glauser M, Saraga E, Vaney AC, Haas R, Kraehenbuhl JP, Blum AL, Michetti P. Oral immunization with Helicobacter pylori urease B subunit as a treatment against Helicobacter infection in mice. Gastroenterology. 1995;109:115–121. doi: 10.1016/0016-5085(95)90275-9. [DOI] [PubMed] [Google Scholar]
- 109.Ikewaki J, Nishizono A, Goto T, Fujioka T, Mifune K. Therapeutic oral vaccination induces mucosal immune response sufficient to eliminate long-term Helicobacter pylori infection. Microbiol Immunol. 2000;44:29–39. doi: 10.1111/j.1348-0421.2000.tb01243.x. [DOI] [PubMed] [Google Scholar]
- 110.Gottwein JM, Blanchard TG, Targoni OS, Eisenberg JC, Zagorski BM, Redline RW, Nedrud JG, Tary-Lehmann M, Lehmann PV, Czinn SJ. Protective anti-Helicobacter immunity is induced with aluminum hydroxide or complete Freund’s adjuvant by systemic immunization. J Infect Dis. 2001;184:308–314. doi: 10.1086/322032. [DOI] [PubMed] [Google Scholar]
- 111.Kipanyula MJ, Seke Etet PF, Vecchio L, Farahna M, Nukenine EN, Nwabo Kamdje AH. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal. 2013;25:403–416. doi: 10.1016/j.cellsig.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 112.Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis. 2008;198:553–560. doi: 10.1086/590158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, Maxeiner J, Hansson M, Taube C, Quiding-Järbrink M, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest. 2012;122:1082–1096. doi: 10.1172/JCI61029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 116.D'Elios MM, Amedei A, Cappon A, Del Prete G, de Bernard M. The neutrophil-activating protein of Helicobacter pylori (HP-NAP) as an immune modulating agent. FEMS Immunol Med Microbiol. 2007;50:157–164. doi: 10.1111/j.1574-695X.2007.00258.x. [DOI] [PubMed] [Google Scholar]
- 117.Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe. 2010;7:265–276. doi: 10.1016/j.chom.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 119.Lu W, Pan K, Zhang L, Lin D, Miao X, You W. Genetic polymorphisms of interleukin (IL)-1B, IL-1RN, IL-8, IL-10 and tumor necrosis factor {alpha} and risk of gastric cancer in a Chinese population. Carcinogenesis. 2005;26:631–636. doi: 10.1093/carcin/bgh349. [DOI] [PubMed] [Google Scholar]
- 120.Rupnow MF, Shachter RD, Owens DK, Parsonnet J. Quantifying the population impact of a prophylactic Helicobacter pylori vaccine. Vaccine. 2001;20:879–885. doi: 10.1016/s0264-410x(01)00401-7. [DOI] [PubMed] [Google Scholar]
- 121.Windle HJ, Kelleher D, Crabtree JE. Childhood Helicobacter pylori infection and growth impairment in developing countries: a vicious cycle? Pediatrics. 2007;119:e754–e759. doi: 10.1542/peds.2006-2196. [DOI] [PubMed] [Google Scholar]
- 122.Shahinian ML, Passaro DJ, Swerdlow DL, Mintz ED, Rodriguez M, Parsonnel J. Helicobacter pylori and epidemic Vibrio cholerae O1 infection in Peru. Lancet. 2000;355:377–378. doi: 10.1016/s0140-6736(99)05143-0. [DOI] [PubMed] [Google Scholar]
- 123.Bhan MK, Bahl R, Sazawal S, Sinha A, Kumar R, Mahalanabis D, Clemens JD. Association between Helicobacter pylori infection and increased risk of typhoid fever. J Infect Dis. 2002;186:1857–1860. doi: 10.1086/345762. [DOI] [PubMed] [Google Scholar]
- 124.de Vries R, Klok RM, Brouwers JR, Postma MJ. Cost-effectiveness of a potential future Helicobacter pylori vaccine in the Netherlands: the impact of varying the discount rate for health. Vaccine. 2009;27:846–852. doi: 10.1016/j.vaccine.2008.11.081. [DOI] [PubMed] [Google Scholar]
- 125.Rupnow MF, Chang AH, Shachter RD, Owens DK, Parsonnet J. Cost-effectiveness of a potential prophylactic Helicobacter pylori vaccine in the United States. J Infect Dis. 2009;200:1311–1317. doi: 10.1086/605845. [DOI] [PubMed] [Google Scholar]
- 126.Velin D, Narayan S, Bernasconi E, Busso N, Ramelli G, Maillard MH, Bachmann D, Pythoud C, Bouzourene H, Michetti P, et al. PAR2 promotes vaccine-induced protection against Helicobacter infection in mice. Gastroenterology. 2011;141:1273–1282, 1282.e1. doi: 10.1053/j.gastro.2011.06.038. [DOI] [PubMed] [Google Scholar]