

Abstract
Abdominal aortic aneurysms (AAAs) are rupture-prone progressive dilations of the infrarenal aorta due to a loss of elastic matrix that lead to weakening of the aortic wall. Therapies to coax biomimetic regenerative repair of the elastic matrix by resident, diseased vascular cells may thus be useful to slow, arrest, or regress AAA growth. Hyaluronan oligomers (HA-o) have been shown to induce elastic matrix synthesis by healthy and aneurysmal rat aortic smooth muscle cells (SMCs) in vitro but only via exogenous dosing, which potentially has side effects and limitations to in vivo delivery towards therapy. In this paper, we describe the development of HA-o loaded poly(lactide-co-glycolide) (PLGA) nanoparticles (NPs) for targeted, controlled, and sustained delivery of HA-o towards the elastogenic induction of aneurysmal rat aortic SMCs. These NPs were able to deliver HA-o over an extended period (>30 days) at previously determined elastogenic doses (0.2 – 20 μg/mL). HA-o released from the NPs led to dose dependent increases in elastic matrix synthesis, and the recruitment and activity of lysyl oxidase (LOX), the enzyme which crosslinks elastin precursor molecules into mature fibers/matrix. Therefore, we were able to successfully develop a nanoparticle based system for controlled and sustained HA-o delivery for the in vitro elastogenic induction of aneurysmal rat aortic smooth muscle cells (EaRASMCs).
Keywords: Nanoparticles, drug delivery, elastic matrix, regenerative matrix repair, hyaluronan oligomers
1. Introduction
Abdominal aortic aneurysms (AAAs), are rupture-prone localized expansions of the aortic wall, which account for over 16,000 deaths a year in the USA [1]. Screening and detection of early stage AAAs in recognized high-risk patients is now possible using high resolution ultrasound, magnetic resonance imaging (MRI), and computed tomography (CT) techniques [2]. Post-diagnosis, AAAs are typically monitored passively every 6 months, during the slow growth phase, until they attain a critical size (>5.5 cm in diameter) or their growth rate exceeds 0.5 cm/year, at which stage surgical intervention is mandated [3]. However, since surgery is unsuitable for many patients and often has high complication rates, a non-surgical AAA treatment, applicable during the slow growth period is desirable [4]. In this context, we seek to develop a matrix regenerative therapy for AAAs. Important to such an approach is the need to stimulate regeneration of elastin and elastic matrix, which unlike collagen, the other major structural component of aortic tissues, is auto regenerated rather poorly in adult tissues.
Elastin is a very important component of connective tissue as it determines the elasticity and resilience, long-range deformability, and passive recoil of the aorta [5]. Auto-regeneration of elastic matrix post-disruption as mentioned, is problematic since, adult and more so, diseased cells are very deficient in elastin synthesis [1]. Accordingly, a significant elastogenic stimulus needs to be provided to coax aneurysmal cells to be able to regenerate disrupted elastic matrix towards stabilizing and potentially arresting AAA growth. Importantly, for net accumulation of de novo synthesized elastic matrix at the AAA site to be achieved, matrix metalloproteinases (MMPs), proteolytic enzymes chronically expressed at the AAA site, must also be attenuated concurrently.
Prior studies in our lab [5] have demonstrated that pro-elastogenic effects of hyaluronan (HA), a matrix glycosaminoglycan (GAG), depending on its molecular weight (MW) or chain length. Oligomeric forms of HA (HA-o), particularly mixtures of HA 4mers and 6mers were capable of providing a significant stimulus for precursor synthesis and elastic matrix assembly [6, 7]. We have previously shown HA-o to induce elastin regeneration by cultured aneurysmal rat aortic smooth muscle cells (SMCs), which are very elastogenically deficient [8]. To ensure that HA-o is delivered in a localized, predictable, and sustained manner to the site of tissue elastolysis, in this study, we investigate the ability of HA-o to be delivered from nanoparticles (NPs) and the effect of surface modification of the NPs on aspects of elastic matrix assembly and proteolysis in vitro within EaRASMC cultures.
2. Methods and Materials
2.1 Formulation of HA-loaded PLGA NPs
Poly(DL-lactide-co-glycolide) (PLGA; 85:15 lactide: glycolide; inherent viscosity 0.55–0.75 dL/g in hexafluoroisopropanol; Durect Corporation, Birmingham, AL) NPs were synthesized, described in a previous paper from our group [9], using a well-established double emulsion solvent evaporation method [10–12]. The NPs were loaded with a mixture of HA oligomers (HA-o) containing 75% w/w of 4mers and 25% w/w 6mers, prepared as described our previous paper [7]. Briefly, PLGA (50 mg) was dissolved in 2 mL of organic solvent, dichloromethane (DCM) or chloroform, depending on the surfactant used. HA-o at desired loadings (3, 5 and 8 wt. % ratios of HA-o:PLGA) was emulsified with the organic solvent by sonication at 20% amplitude. The aqueous phase consisted of 12 mL of nanopure water, containing polyvinyl alcohol (PVA) or didodecyldimethylammonium bromide (DMAB) as the surfactant at a concentration of 0.25% w/v. Chloroform was used as a solvent with DMAB and DCM with PVA. The organic phase was added to the aqueous phase and sonicated, then left stirring overnight to evaporate the organic phase. Samples were desiccated for 1 hour under vacuum prior to NP separation by ultracentrifugation at 35,000 rpm for 30 minutes (Beckman L-80, Beckman Instruments Inc., Palo Alto, CA). After separation, the NPs were further washed twice with nanopure water, sonicated, and centrifuged at 30,000 rpm for 30 minutes prior to lyophilization (48 hours) to generate NPs in a sterile dry powder form. The supernatants from the separation and wash steps were analyzed to calculate HA-o loading efficiency within the NPs.
2.2 Determination of size and surface charge of HA-o loaded PLGA nanoparticles
The mean hydrodynamic diameters of the NPs were determined by dynamic light scattering and NP surface charges or ζ-potentials were estimated with phase analysis light scattering, as described earlier [9]. Both measurements were performed on a commercial particle-sizing system (PSS/NICOMP 380/ZLS, Particle Sizing Systems, Santa Barbara, CA).
2.3 Isolation of healthy and aneurysmal rat aortic SMCs
Animal procedures were all conducted with approval of the Institutional Animal Care and Use Committee (IACUC) at the Cleveland Clinic. The animal facility is AAALAC-approved and has animal assurance (#A3145-01; Expires 04/30/15). Aneurysmal rat aortic SMCs (EaRASMCs) were isolated from AAAs generated in adult male Sprague-Dawley rats (n = 3) via elastase-infusion, as previously described by our lab [8, 13], at 14 days post-induction. Briefly, the AAA tissue was harvested, cut longitudinally and the intimal layer was scraped off, the medial layer then dissected from the adventitial layer and chopped into ~0.5 mm slices and rinsed twice in 37 ºC PBS. The tissue pieces were enzymatically digested in DMEM-F12 medium (Invitrogen, Carlsbad, CA) containing 125 U/mg collagenase (Worthington Biochemicals, Lakewood, NJ) and 3 U/mg elastase (Worthington Biochemicals) for 30 minutes at 37 ºC. These were then centrifuged at 400 g for 5 minutes and cultures in T-75 flasks for ~ 2 weeks in DMEM-F12 medium (Invitrogen) supplemented with 10% v/v fetal bovine serum (FBS; PAA Laboratories Inc., Etobicoke, Ontario) and 1% v/v penicillin-streptomycin (PenStrep; ThermoFisher, South Logan, UT). The isolated primary EaRASMCs were propagated for 2 weeks and passaged when they reached confluence.
2.4 Cytotoxicity of PLGA NPs and evidence of infiltration or exclusion of NPs by cells
The dose-dependent cytotoxic effects of the PLGA NPs generated using DMAB and PVA as stabilizers, were quantified using a LIVE/DEAD® viability assay (Invitrogen). According to the manufacturer, this assay has been validated by several laboratories as a faster, safer and more sensitive method indicating cytotoxicity, compared to other methods. Additionally, the assay has been used to quantify cell death via apoptosis [14, 15] and cell-mediated cytotoxicity and necrosis [16, 17]. Passage 2 EaRASMCs were seeded at a concentration of 7.5 × 104 cells/well in sterile 6-well plates (Area = 9.6 cm2) and cultured in DMEM-F12 cell medium supplemented with 5% v/v FBS and 1% v/v PenStrep for 48 hours. PLGA NPs prepared with either PVA or DMAB were added to the cells at different concentrations (0, 0.2, 0.5, and 1 mg/mL) and the cells further cultured for 7 days. Thereafter, the cells were assayed and imaged on a fluorescent microscope (IX51, Olympus America, Center Valley, PA), with five random regions being visualized per replicate culture.
2.5 HA-o loading efficiency and release from PLGA NPs
A hyaluronan carbazole assay was used to detect and quantify HA-o release from NPs. The assay is based on the established modified Bitter-Muir carbazole method [18]. To determine the loading efficiency of HA-o within the NPs, supernatant saved from their synthesis were filtered in a 3kDa filter centrifuge tube (Amicon, Billerica, MA) and assayed for HA content. The release study was carried out using a double chamber 3 kDa filter centrifuge tube, which was loaded with 8 mL PBS in the receiver side and 3 mL of NP suspension in phosphate buffered saline (PBS) on the donor side; 0.5 mL aliquots were removed from the receiver chamber and replaced with fresh PBS at each time point (days 0, 1, 4, 7, 11, 14, 17, 21, 26, & 31). The tubes were incubated on a shaker in a 37 °C chamber to simulate physiological conditions and keep the nanoparticles in suspension. Each 0.5 mL sample was assayed for HA-o content and compared with known standards (4 and 6 mer mixture of known concentrations) in order to quantify the extended release profiles of HA-o from the NPs. Based on a known volume (0.5 ml) of sample removed and the HA-o concentration measured within, the (a) amount of HA-o released since the previous sampling event and (b) cumulative release of HA-o up to that time point, were calculated using the following formulae: Xn+1 = Xn+1 – Xn*(10.5/11) and Xtotal=X1+X2+…Xi, where ‘X’ represents the amount of HA-o detected in the sampled volume, and ‘n’ represents the sampling event itself. The loading efficiency was calculated based on the unencapsulated HA detected in the supernatant using the formula 100 × [(loaded HA-unencapsulated HA)/loaded HA].
2.6 Cell culture with nanoparticles
Passage 2 EaRASMCs were seeded at 6 × 104 cells/well in 6-well plates and 3 × 104 cells/well in 2-well chamber slides (Area = 4.2 cm2) and cultured for 72 hours in DMEM-F12 medium supplemented with 10% v/v FBS and 1% v/v PenStrep. After this period, the medium was aspirated and replaced with an NP suspension prepared in fresh medium containing 5% v/v FBS and 1% v/v PS. These suspensions contained NPs loaded with either 0 (control), 3, 5, or 8% w/w HA-o provided at a concentration of 0.5 mg/mL (n = 6 cultures per HA-o loading). EaRASMCs cultured with no NPs served as additional treatment controls. The cells were cultured for 21 days. During each medium change, the spent medium was removed and centrifuged at >10,000 rpm for 30 minutes in a microcentrifuge (Beckman Microfuge 16®, Beckman-Coulter, Inc.) to spin down the NPs. After aspirating the supernatant, the NPs were re-suspended in fresh medium, and then added back to cell layers. At day 21 of culture, each cell layer was harvested in the appropriate buffer for each assay and chamber slides were fixed for immunofluorescence and transmission electron microscopy (TEM).
2.7 DNA assay for cell proliferation
DNA content of the cell layers was measured using a fluorometric assay to determine the increase in cell number so as to quantify cell proliferation [19]. Quantified elastin matrix amounts in cell culture were normalized to their respective DNA contents to provide a reliable basis for comparison between replicate cultures and treatments. Briefly, cell layers at day 1 and 21 of culture were harvested in 1.0 mL of Pi buffer (50 mM Na2HPO4, 2 mM EDTA, 0.02% w/v NaN3) and 100 μL samples were assayed. Cell counts were calculated based on an estimated of 6 ng DNA per cell [19].
2.8 Fastin assay for elastin
The Fastin Assay (Accurate Scientific and Chemical, Westbury, NY) was used to quantify the total elastin content in the cell layers including alkali-soluble and insoluble fractions based on our previously described protocol [20, 21]. Cell layers at day 21 of culture were harvested in 1.0 mL of Pi buffer and sonicated to homogenize it uniformly. The homogenized cell layer was digested with 0.1 N NaOH for 1 hour at 95 °C and centrifuged to yield a densely packed pellet of insoluble elastin, with the alkali-soluble portion remaining in the supernatant, which was then stored for subsequent analysis. As the Fastin Assay only quantifies soluble α-elastin, the insoluble elastin pellet was solubilized using 0.25 M oxalic acid for 1 hour at 95 °C. After the solubilization of this alkali-insoluble fraction the samples were pooled and centrifuge-filtered using microcentrifuge tubes with a 10 kDa molecular weight cut-off (Millipore, Billerica, MA). These soluble and insoluble fractions were measured with the assay and quantified using standards of know elastin content. The elastin content for each treatment condition was normalized to its corresponding DNA content for accurate comparison between the test conditions.
2.9 Lysyl oxidase (LOX) activity assay
An Amplex Red Assay (Invitrogen) was used to determine LOX activity in cell layers harvested after 21 days of culture in harvested in RIPA buffer (Thermo Scientific) containing Halt™ protease inhibitor (Thermo Scientific). Samples were assayed for total protein content using a bicinchonic acid (BCA) assay kit (Thermo Scientific). Briefly, 50 μL of samples from cell culture medium or cell layers was mixed with 50 μL of a 50 mM sodium phosphate reaction buffer (pH 7.4) containing 1.2 M urea, 10 mM 1,5-diaminopentane (Sigma–Aldrich), 0.2 U ml−1 horseradish peroxidase (HRP; Invitrogen) and 10 μM Amplex® Red (Invitrogen). Control samples (50 μL) were mixed with sodium phosphate reaction buffer containing only 0.2 U ml−1 HRP and 10 μM Amplex® Red. Peroxide standards were used to generate a calibration curve and control sample absorbances were subtracted from their test case samples and then normalized to sample protein content. The assay measures the hydrogen peroxide released from the deamination of the diamino compounds by lysyl oxidase.
2.10 Western blots for MMP-2 and LOX protein synthesis
Protein synthesis of elastolytic MMP-2 by EaRASMCs was semi-quantitatively compared across treatments using western blots. EaRASMC layers at day 21 of culture were harvested in 0.5 mL of RIPA buffer containing HALT protease inhibitor (Thermo Scientific). The cell layers were assayed for total protein content with a BCA assay kit (Thermo Scientific) to determine optimal protein loading for the blots. Maximum loading conditions were used for each sample under reduced conditions. Samples were run in a 10% sodium dodecylsulfate-polyacrylamide electrophoresis (SDS-PAGE) gel (Invitrogen) for MMP-2 and a 4–12% gel was used for LOX, a BenchMark™ pre-stained molecular weight ladder (Invitrogen) was also run in addition to a MMP-2 and LOX standard. Gels were transferred onto nitrocellulose membrane (iBlot Western Blotting System, Invitrogen). After 1 hour of blocking with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE) the gels were immunolabeled for 16 h at 4 ºC with a rabbit polyclonal antibody against MMP-2 (1:500 dilution; Abcam, Cambridge, MA) and a rabbit monoclonal antibody against LOX (1:200 dilution; Abcam), with a mouse monoclonal antibody against β-actin (1:1000 dilution; Sigma-Aldrich) as the loading control. Secondary antibodies labeling occurred for 1 hour at room temperature using IRDye 680LT goat-anti-rabbit (1:15,000 dilution) and IRDye 800CW goat-anti-mouse (1:20,000 dilution) polyclonal antibodies (LI-COR Biosciences). A LI-COR Odyssey laser-based scanning system was used for the fluoroluminescent detection of protein bands. Band intensities for MMP-2 and LOX were quantified using the ImageJ software for each test case and normalized to its corresponding β-actin control band intensity for proper comparison between test cases.
2.11 Immunofluorescence detection of elastic matrix assembly proteins
EaRASMC layers were cultured on two-well Permanox® chamber slides (Nalge Nunc International, Penfield, NY) for 21 days, the cultures rinsed twice with PBS and fixed with 4% w/v paraformaldehyde and treated with rabbit anti-rat primary polyclonal antibodies against Elastin (Millipore, Billerica, MA), and finally visualized with Alexa633-conjugated IgG secondary antibodies 1:1000 v/v (Invitrogen). EaRASMCs cultured with no NPs were labeled as the treatment control and cells with no primary antibodies served as the immuno-labeling controls. Vectashield® mounting medium containing the nuclear dye, 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories; Burlingame, CA) was used during mounting to label cell nuclei. To add a spatial component to the analysis of the cell bodies, the filamentous actin was fluoro-labeled with Alexa488 Phalloidin at a 1:50 dilution (Invitrogen). Imaging for 3–D analysis to determine the location and structure of the elastin confocal microscopy was carried out on a TCS SP5 II microscope (Leica Microsystems, Inc., Buffalo Grove, IL); z-stack maximum projections were created in software based on overlays of the 1 μm sections across the thickness of the cell layer.
2.12 Transmission Electron Microscopy (TEM)
TEM was used to characterize the ultrastructure of the elastic matrix generated by cells in presence and absence of HA-o loaded NPs. EaRASMCs were cultured for 21 days on 2–well chamber slides were washed twice with cell medium followed by a wash with PBS (37 °C). Cell layers were then fixed first for 5 minutes at 37 °C, and then at 4 °C overnight with 4% v/v paraformaldehyde / 2.5% v/v Glutaraldehyde in 0.1 M sodium cacodylate buffer. The samples were post-fixed in 1% w/v osmium tetroxide (1 hour), dehydrated in a graded ethanol series (50–100% v/v), embedded in Epon 812 resin, sectioned, placed on copper grids, stained with uranyl acetate and lead citrate, and imaged on a Philips CS12/STEM (FEI Company, Hillsboro, OR).
2.13 Statistical Analysis
All experimental data are presented at n = 6 per condition unless otherwise stated as a mean ± standard deviation (SD). Statistical significance was evaluated with a student’s t-test with p < 0.05 considered as being statistically significant.
3. Results
3.1 Formulation and Size & Charge Characterization of HA-o loaded PLGA NPs
The NPs formulated with 0.25% w/v of PVA and DMAB exhibited hydrodynamic sizes of 376.5 ± 35.8 nm and 309.8 ± 15.3 nm and ζ-potentials of −29.3 ± 7.9 mV and +44.8 ± 3.5 mV respectively. Incorporation of HA-o at different loading percentages did not alter the size and charge of the NPs (Table 1).
Table 1.
Size and surface charge of 85:15 PLGA NPs. (n = 6 replicates, mean ± SD).
| Surfactant | HA Loading | Size [nm] | ζ-Potential [mV] |
|---|---|---|---|
| 0.25 % PVA | 0% | 376.5 ± 35.8 | −29.3 ± 7.9 |
| 3% | 336.5 ± 10.4 | −27.0 ± 5.4 | |
| 5% | 304.9 ± 9.9 | −32.6 ± 4.5 | |
| 8% | 315.2 ± 9.0 | −28.1 ± 0.5 | |
|
| |||
| 0.25 % DMAB | 0% | 309.8 ± 15.3 | +44.8 ± 3.5 |
3.2 Cytotoxicity of NPs based on concentration and surfactant
As seen in the representative images included in Figure 1, PLGA NPs surface modified with PVA did not adversely impact viability of EaRASMCs, over 7 days of culture when provided to the cells at NP doses of 0.2, 0.5, and 1.0 mg/mL, although a few dead cells (red) were seen in both NP-free (control) and blank PVA NP-treated cultures. Differently, DMAB NPs appear to be mildly cytotoxic at a concentration of 0.5 mg/mL and were found to significantly reduce viability when exposed to cells at a concentration of 1.0 mg/mL; in the latter case, no viable cells were observed.
Figure 1.
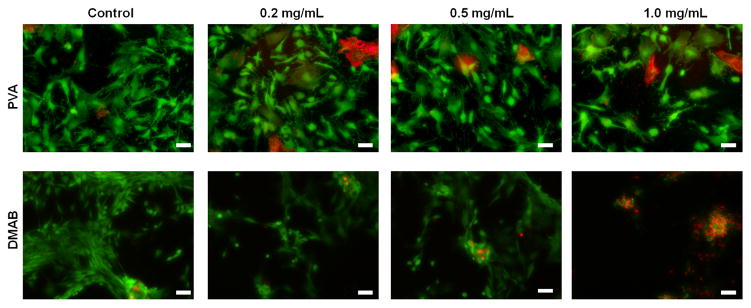
Representative images illustrating the viability of rat aneurysmal SMCs (EaRASMCs) upon exposure to HA-o loaded (3, 5 and 8% w/w) and unloaded (0%) PLGA NPs prepared using PVA and DMAB (0.25 % w/v concentration) as stabilizers. The LIVE-DEAD® assay stains live cells green and dead cells red. (Scale bar equivalent to 100 μm).
3.3 Efficiency of HA-o encapsulation efficiency in PLGA NPs and release kinetics
The encapsulation efficiencies for HA-o loaded into the PVA-modified PLGA NPs at 3, 5, and 8% w/w loading were 85.9% ± 4.7%, 80.1% ± 4.7%, and 86.7% ± 5.8% respectively (n = 3 / formulation; mean ± SD). For all NP concentrations and HA-o percent loadings tested, controlled release of HA-o was maintained through the 31 days of sampling (Figure 2). As the NP concentration in solution increased, both the initial HA-o release rate and the cumulative % of HA-o released, decreased. As the HA-o loading % increased the initial burst release rate increased, however, the cumulative percent of loaded HA-o released decreased. Even so, the 8% loading was able to deliver the largest dose of HA-o though it had a plateau for the release at around 15% starting day 7 while the 3 and 5% had better extended release though delivered smaller, yet still elastogenic doses of 20–80 μg based on prior studies [22–24] (Figure 2). Overall, for all test conditions (HA-o loadings and NP concentrations; Figure 2), we observed that significant release of HA-o occurred over the first 14 days, after which the net amount of HA-o newly released at the successive time points assayed thereafter, decreased as a result of which the release curves appear to plateau. However, as shown in Supplementary Table 1, the HA-o released from the NPs between the time points during this period was quite significant, representing ≥ 2% of the total HA-o released, and thus confirming that release of HA-o from the NPs continued at these later time points of our release study.
Figure 2.

In vitro release profiles of HA-o from PVA-modified PLGA NPs in 1× phosphate buffered saline (PBS), pH 7.4. The NPs were loaded with HA-o at 3%, 5%, and 8% w/w HA-o to PLGA, and delivered at three different NP concentrations (0.2, 0.5, and 1.0 mg/mL) (n = 3 per group; mean ± SD). (A) HA-o released as a percent of the total encapsulated HA-o based on encapsulation efficiency data (B) absolute concentration of released HA-o in PBS.
3.4 Cell proliferation
There was no significant difference in cell proliferation ratios (i.e., fold changes in cell numbers at 21 days vs. 1 day of culture) between the NP-free & NP-treated (blank or HA-o loaded) cultures as seen in Figure 3. The increase in cell number, rather than a decrease, over the culture period, in cultures supplemented with HA-o releasing NPs were consistent with the lack of cytotoxic effects at the provided dose (0.5 mg/mL; see section 3.2).
Figure 3.
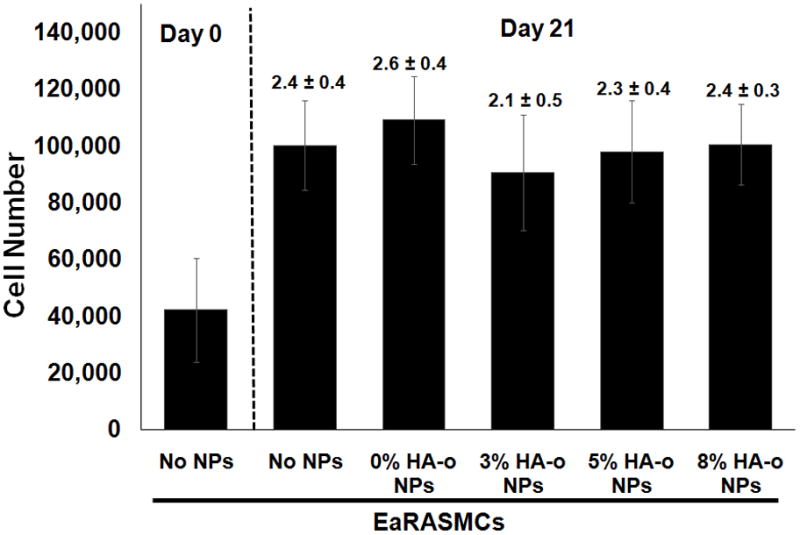
Proliferation data for EaRASMCs in response to HA-o released from PVA-NPs at three different HA-o loadings (3%, 5%, and 8%). The NP concentration in all cases was 0.5 mg/mL. Values above error bars indicate fold increases in mean ± SD of cell numbers vs. day 1 of the culture experiment. EaRASMCs cultured standalone served as the treatment control and cells cultured with blank NPs (0% w/w DOX) were regarded as the active agent control. The cell number was calculated based on an estimate of 6 pg of DNA per cell, via a DNA assay, at 1 and 21 days post-seeding (All values are mean ± SD; n = 3 per case).
3.5 Quantification of elastic matrix
A Fastin assay was used to quantify cell deposited elastic matrix, including both the alkali-soluble fraction and more robustly cross-linked alkali-insoluble fraction. Over a 21 day period, NP-untreated EaRASMCs generated a total of 32.1 ± 3.4 μg/well of matrix elastin. Culture of EaRASMCs with blank (0% HA-o) and 3% w/w HA-o loaded NPs did not influence elastin matrix synthesis versus no NP control cultures on a per cell basis (Figure 4). However, NPs loaded with 5% w/w and 8% w/w HA-o were found to significantly increase the total elastin matrix synthesis on a per cell basis (p < 0.05) compared to cultures that received blank NPs. The cultures that received NPs containing 8% w/w HA-o yielded both alkali-soluble & alkali insoluble elastic matrix fractions that were increased significantly over no NP controls on a per cell basis (1.36 ± 0.11–fold and 1.28 ± 0.06-fold increases vs. untreated EaRASMCs respectively; p = 0.04 and p = 0.03).
Figure 4.

Fold-differences in DNA-normalized amounts of alkali-soluble, insoluble, and total elastic matrix synthesized by EaRASMCs in response to blank PVA-NPs and HA-o released from NPs, compared to NP-untreated EaRASMC cultures. Results indicate mean ± SD amounts measured from a total of n = 6 replicate cultures/case; * denotes p < 0.05 compared to NP-untreated EaRASMC controls.
3.6 LOX protein synthesis and enzyme activity
Figure 5A shows differences in LOX protein synthesis, as measured via western blots in EaRASMC cultures with no NPs, blank NPs, and NPs containing different loadings of HA-o at a concentration of 0.5 mg/mL. While the blank NPs and the NPs loaded with 3% w/w HA-o did not enhance LOX protein synthesis relative to NP-untreated cultures, NPs loaded with 5% and 8% w/w of HA-o, significantly enhanced LOX protein synthesis relative to NP-untreated cultures (2.5 ± 0.3-fold and 2.7 ± 0.2-fold increases with p = 0.03 and p = 0.02 respectively vs. EaRASMCs alone). The Amplex Red assay was used to compare activities of cell-layer-associated LOX enzyme in EaRASMC cultures, across treatments. As seen in Figure 5B, at the delivered dose of 0.5 mg/mL, both blank NPs and those loaded with HA-o significantly increased LOX enzyme activity relative to NP-free EaRASMC cultures (p < 0.05). NPs containing the highest HA-o loading (8% w/w) induced the greatest increases in LOX activity (2.7 ± 0.2 fold vs. no NP controls; p = 0.02) which was also deemed to be significantly higher than that in the blank NP cultures (1.6 ± 0.2 fold increase; p = 0.04 vs. blank NP condition).
Figure 5.
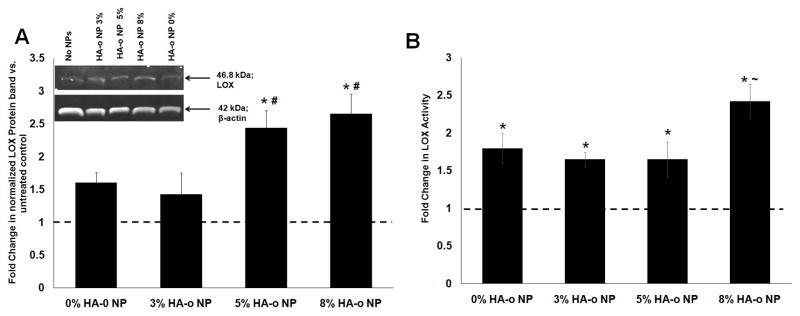
Effects of PVA-NPs and HA-o released from NPs on LOX (A) protein synthesis and (B) enzymatic activity. (A) Fold change in LOX protein synthesis by EaRASMC cultures treated with HA-o loaded (0, 3, 5 and 8% w/w) PLGA NPs relative to NP-untreated EaRASMC controls. LOX band intensity for each sample was normalized to intensity of the corresponding β-actin band and then further normalized to similarly calculated values for the control cultures. Inset: representative image of a western blot illustrating the expression of LOX protein, along with β-actin bands as loading controls. (B) Fold-change in LOX activity at 21 days in EaRASMC cultures treated with NPs containing different levels of HA-o loading. LOX activity was measured in the cell layer by the quantity of hydrogen peroxide released upon oxidative deamination of alkyl monoamines and diamines, normalized to that of the NP-untreated controls. * denotes p < 0.05 compared to EaRASMCs; # denotes p < 0.05 compared to 0 and 3% HA-o loaded NPs; ~ denotes p < 0.05 compared to 0, 3, and 5% HA-o loaded NPs. (Data represented as mean ± SD; n = 3 per case for Western blots, n = 6 per case for LOX activity assay; * denotes p < 0.05 compared to controls with no NPs). The normalized band intensity of LOX for the NP-untreated control test condition was set to unity to determine the fold change in LOX expression or activity for the different NP-treated test cases.
3.7 Effect of HA-o released from PLGA NPs on MMP-2 production
The zymogen band for MMP-2 was undetectable in our western blots. There was no significant change in production of active MMP-2 when EaRASMCs were treated with blank PVA-modified NPs, or PVA-NPs loaded with HA-o relative to NP-free controls (p > 0.05) (Figure 6).
Figure 6.

Effect of HA-o released from NPs on MMP-2 synthesis in EaRASMC cultures, as analyzed with Western blots. (A) Representative image of Western blot, with β-actin bands as loading controls. (B) Fold change in production of active MMP-2 compared to control cultures without NPs. Data represented as mean ± SD; n = 3 per case * denotes p < 0.05 compared to controls with no NPs. Note: the values in (B) represent the fold change in normalized band intensities of the active MMP-2 band for each treatment test case (normalized to its corresponding β-actin loading control band to enable accurate comparison between the different test cases) compared to untreated (no NP) controls. The normalized band intensity of MMP-2 for the NP-untreated control test condition was set to unity to determine the fold change in MMP-2 expression or activity for the different NP-treated test cases.
3.8 Immunofluorescence detection of elastin
As seen in representative images higher magnification (40×) confocal microscopy images in Figure 7, most of the deposited elastin (pink) was localized in the extracellular space. As seen in this image panel, elastic fiber like structures are seen in cultures that received NPs containing the highest loading of HA-o (8% w/w HA-o to PLGA). In cell layer with 3% HA-o loaded NPs there was a greater amount of elastin detected over the cell layer with 0% HA-o loaded NPs, however, it did not exhibit the same fiber like structures seen in the 8% HA-o loaded NP cultures.
Figure 7.

Confocal micrographs showing 21-day EaRASMC layers cultured with blank (active agent controls) and HA-o-loaded PVA-NPs. The cultures were immunofluorescence- labeled for elastin (false-colored magenta; white arrows), which is mostly localized extracellularly. Nuclei were stained with DAPI (blue) and filamentous actin was stained with Alexa Fluor 488 phalloidin (green).
3.9 Evaluation of elastin ultrastructure via transmission electron microscopy
Compared to EaRASMCs cultured with no NPs, which contained only a few sporadic deposits of amorphous elastin (Figure 8A), EaRASMC layers cultured with HA-o NPs (shown here for the 8% w/w HA-o loaded NPs) contained significantly greater number of amorphous elastin deposits which in several locations appeared to coalesce and form elongated agglomerates evocative of nascent elastic fiber formation (Figures 8B & C). Importantly, the amorphous elastin deposits in the NP-supplemented cultures were closely associated with abundant peripheral microfibrils (Figures 8C & D).
Figure 8.

Transmission electron micrographs illustrating the effects of HA-o released from NPs loaded with 8% w/w HA-o over 21 days on elastic matrix deposition (B, C, D) by EaRASMCs, compared to NP-untreated control (A). E indicates cross sections of immature amorphous elastin deposits in the fibrillar network, and M indicates the microfibrillar scaffold structure surrounding the deposits.
4. Discussion
Elastin, along with collagen, is important structural ECM proteins. Collagen, which primarily imparts stiffness to soft tissues, is generated prolifically by cells (SMCs, fibroblasts) well into adulthood and in response to injury [25]. However, adult cells are inherently poor in their ability to synthesize elastin and crosslink it into a mature matrix, although this can be overcome by exogenous delivery of elastogenic biomolecules [26], Therefore, our long-term goal is to develop modalities for localized, controlled, and sustained delivery of HA-o to tissue sites of proteolytic disease such as within AAA tissues towards inducing biomimetic cellular regeneration of the elastic matrix, which poorly occurs naturally.
In previous work, we identified specifically-sized HA oligomers that elastogenically stimulate adult SMCs without any observable changes to their phenotype [7, 27]. However, HA oligomers of certain sizes can also induce inflammatory effects under specific tissue micro-environmental conditions, especially if delivered in an uncontrolled and systemic manner [28, 29]. In this context, HA-o delivery using polymeric carrier vehicles can offer considerable benefit over systemic delivery of active agents [30]. Using nanoparticles (NPs) as drug delivery vehicles proffers several advantages over the use of microparticles, due to their higher surface area to volume ratio which greatly enhances release rates of active agents and their generation of smaller amounts of potentially cytotoxic polymer degradation products. In the case of polymers such as PLGA, this will ensure that the pH is not lowered via the accumulation of acidic byproducts. Additionally their small size would potentially enhance their uptake and retention within the vessel/aortic wall [10, 11].
The NPs in this study were formulated using PLGA as it has been approved by the FDA for drug delivery applications in humans due to its biodegradability, biocompatibility, and non-toxicity [31]. PLGA is a copolymer of poly(lactic acid) (PLA) and poly(glycolic acid) (PGA). For this application seeking to achieve extended release of the HA-o from NPs, we selected a copolymer weight ratio of 85:15 PLA to PGA, since it has been shown that increasing the lactide to glycolide ratio decreases the polymer degradation rate and therefore can slow the release of incorporated active agent [32]. We selected a high molecular weight (~87 kDa) form of the polymer to create NPs with the expectation that it would prolong duration of release of active agents since it has been shown that polymers of higher molecular weight degrade more slowly [33].
Size and surface charge of the NPs also play an important role in their in vivo efficacy. In a recent study by our group [9], we demonstrated that PLGA NPs of 100 and 200 nm sizes were internalized by EaRASMCs, while those of 500 nm size were localized in the extracellular space. However, NPs > 500 nm in size have been shown to induce phagocytosis [34], while those < 100 nm elicit an immune response [35]. The size of ~350 nm that we obtained for our NP preparations is ideal for our purpose, as NPs of this size have been predominantly shown to localize in the extracellular space, where elastic matrix assembly occurs [9]. The zeta potential of the NP surface is also a critical determinant of their colloidal stability, with NPs exhibiting a zeta potential of magnitude > 30 mV being considered to be colloidally stable in suspension [36, 37]. The NP preparations we generated had zeta potentials of −30 mV for PVA and +33 mV for DMAB, confirming their stability in suspension.
Our results indicated that PVA surface-modified NPs supplemented to EaRASMC cultures did not have any significant adverse impact on cell viability relative to NP-free control cultures, at provided NP doses up to 1 mg/mL. Different from the PVA NPs, DMAB-modified NPs were cytotoxic at doses >0.5 mg/mL. This could be due to two possible reasons. Studies have shown that positively charged NPs electrostatically interact with the negatively charged cell surface and are internalized readily into cells by endocytosis, unlike negatively charged NPs [38]. Further, accumulation of positive charged NPs within the cell has been shown to trigger influx of chloride ions into the cell to maintain charge neutrality, resulting in osmotic swelling and cell rupture/death [39]. This might have accounted for the reduced viability of EaRASMCs when cultured with the higher doses of DMAB-modified PLGA NPs. Thus, although a positive surface charge would enhance NP uptake into the aortic wall multifold [11], on account of cytotoxic effects observed at increased concentration of DMAB-modified NPs, all subsequent studies were conducted with the PVA-modified NPs alone.
PVA is the most commonly used stabilizer in formulating PLGA NPs. NPs formulated with PVA demonstrate reduced cellular uptake due to hydrophilicity of the NP surface [31], as well as the negatively charge on the cell surface. This is beneficial for our proposed application, since it would enable released active agents to interact closely with their cell-surface receptors (e.g., CD44 for HA-o) and through the auspices of these released agents and also the physical characteristics of the NPs, influence elastic matrix assembly, which occurs in the extracellular space. In addition, PVA is known to increase encapsulation efficiency of water-soluble agents due to viscosity of the PVA resisting the outward diffusion from the internal aqueous phase [40]. The encapsulation efficiencies of HA-o for all loadings were 80-90% indicating that it was efficiently encapsulated.
Our data indicated that PVA-NPs are suitable for controlled and sustained release of HA-o for more than 30 days at physiological pH (Figure 2). Although a burst release of HA-o was initially observed from the NPs under all tested loading conditions, prior to attaining a plateau, the HA-o released during this phase, which lasted ~1 day, accounted for only ~5% of the HA-o loaded. While as expected, we observed absolute amounts of HA-o released from the PVA-NPs, for any specified NP concentration, to positively correlate to percent HA-o loading, we also observed a steady decrease in the cumulative fraction of encapsulated HA-o that was released. As the HA-o percent loading increased there was a decrease in the overall percent of encapsulated HA-o that was released from >30% for the 3% HA-o loading to ~20% for the 8% HA-o loading at 30 days. This observation is consistent with our prior findings with DMAB-modified PLGA NPs releasing doxycycline [9], and is possibly due increased packing of HA-o molecules within the PLGA matrix with higher % HA-o loading resulting in reduced infiltration of aqueous medium, and as a result a reduction in the fraction of HA-o that dissolves and diffuses out. Since the absolute concentration of released HA-o in solution is higher for higher NP concentrations, it is likely that the concentration gradient of HA-o between the inside and the outside of the NP would have been reduced under these conditions, resulting in a reduced driving force for outward diffusion of HA-o.
We found HA-o released from PVA-modified NPs to have no net effects on cell viability or proliferation, relative to NP-free control cultures (Figure 3), similar to our previous observations with exogenous HA-o [41]; active proliferation of the cells indicated the absence of any cytotoxic effects due to the HA-o or NPs. The absence of any attenuatory effects on cell proliferation by blank PVA-modified NPs relative to NP-free cultures confirmed that neither PLGA nor PVA, at the 0.5 mg/mL NP dose, affect cell viability.
Hyaluronic acid (hyaluronan, HA) a glycosaminoglycan component of the extracellular matrix (ECM) [30] has been suggested to play an indirect but vital role in elastin precursor recruitment and fiber assembly through its association with versican and also due to its highly anionic nature which encourages opposite charge interactions with tropoelastin molecules [42]. We have previously shown HA-o to induce elastogenesis in vitro when administered exogenously to vascular smooth muscle cells [5–7, 21, 41, 43]. In an earlier publication, we showed HA-o in the dose range of 0.2 to 20.0 μg/mL to elastogenically stimulate rat aortic SMCs [8]. However, in this earlier work, the culture medium was periodically replenished, with addition of a fresh bolus of exogenous HA-o. This approach is associated with fluctuations in bioavailability of HA-o due to its depletion in cell culture and periodic replenishment. Moreover, delivery of exogenous HA-o to tissues in vivo is unlikely to be useful due to (a) relatively short half life of HA-o in the bloodstream [44], (b) limitations to diffusion of HA-o to the interior of the tissue, resulting in a spatial concentration gradient and thus, non-homogenous cell responses within the tissue space. The encapsulation of HA-o within PLGA NPs would not only enhance its circulatory half-life, but also enable its localized delivery at the desired site. The ability to achieve controlled and sustained release of equivalents of the previously identified initial elastogenic doses of HA-o from NPs will thus be highly beneficial. In this study, we have been able to release HA-o in a controlled manner from NPs, especially those loaded with 3 or 5% w/w HA-o and delivered at NP concentrations of 0.5 mg/mL, to generate steady state concentrations in aqueous solutions that lie well within the elastogenic low μg/ml dose range.
Delivery of HA-o from 5% w/w and 8% w/w HA-o loaded NPs was found to significantly augment cellular deposition of elastic matrix relative to blank NP-supplemented control cultures (Figure 4), with 8% w/w HA-o loaded NPs inducing increases in both the alkali-soluble and highly crosslinked alkali-insoluble elastin fractions. Although we did not measure tropoelastin precursor synthesis in this study, based on our prior studies which have shown exogenous HA-o to enhance tropoelastin synthesis [1, 5, 23, 41], this increased elastic matrix deposition observed in this study may be due to increased production of elastin precursors upon stimulation with HA-o.
LOX is a copper-dependent amine oxidase that plays an important role in the stability of the elastic matrix by catalyzing the crosslinking of tropoelastin into mature elastic fibers [45]. In our study we found a significant increase in LOX production in all cultures supplemented with NPs and LOX synthesis for both the 5% and 8% w/w HA-o loadings but not with the blank NPs or NPs loaded with 3% w/w HA-o. The increase in LOX production is consistent with results of prior studies wherein we showed desmosine crosslinking of elastin to be significantly up-regulated in exogenous HA-o supplemented cultures versus HA-o-free cell cultures [7, 8]. Thus, increased crosslinking of elastin precursors is at least in part, responsible for the increased deposition of elastic matrix in 5% w/w and 8% w/w HA-o NP-supplemented cultures. This is an important outcome since an effective matrix regenerative therapy to reverse the effects of elastolytic disruption within AAA requires formation of new mature elastic matrix for which LOX-mediated crosslinking is vital. In this regard, recent studies have (a) shown ECM degradation in rats with elastase-induced AAAs to correlate with decreased LOX expression [46], emphasizing the importance of LOX to allowing or reversing AAA progression, and (b) generated evidence in mice with AAAs that overexpression of LOX does slow AAA growth [47].
The increase of LOX activity in cultures with blank PVA-NPs over control cultures was surprising, considering that previous findings in our lab showed LOX activity to moderately decrease in the presence of blank PVA-NPs vs. NP-untreated controls [9]. To explain this, we evoke previous work by Weissen-Plenz et al., who demonstrated that decreases in tropoelastin mRNA expression lead to concomitant reduction in LOX and bone morphogenetic protein-1 (BMP-1) mRNA levels in vascular SMC cultures [48]. Since BMP-1 mediates the proteolytic conversion of LOX from its zymogen form to its active form [49], we therefore hypothesize that in our cultures, the increases in both elastin (Figure 4) and LOX protein content (Figure 5A) is accompanied by upregulated BMP-1 production and/or activity, which would in turn result in augmented conversion of LOX from its proenzyme form to its active form. This could potentially explain the enhanced LOX activity observed in Figure 5B for the HA-o treated cultures. Future studies will seek to elucidate the exact mechanisms underlying the activation of LOX and BMP-1 due to PVA functionalized NPs and the released HA-o.
Hyaluronan can induce significant increases in cellular MMP-2 and MMP-9 production in vitro under specific circumstances dictated by cell type, cellular microenvironment and health, and HA size [50, 51]. Our study demonstrated no significant differences in MMP-2 synthesis between the control cultures or blank PVA-NPs and HA-o loaded PVA-NPs, which is likely due to the low steady state aqueous concentrations of HA-o released from NPs, This would be highly desirable from the standpoint of AAAs which are characterized by chronic overexpression of MMPs. However, we acknowledge that in vivo, other cells (particularly inflammatory cells) would also produce MMPs. Thus, based on the results from this study, which indicate that these HA-o NPs are unlikely to actively provide benefit in terms of attenuating MMP production and activity, co-delivery of MMP-inhibitors along with HA-o NPs may be required to preserve regenerated matrix within AAAs. Hence, our future work in vivo will provide insights into the ability of these HA-o loaded NPs to potentially avoid increased cellular production of MMPs.
Immunofluorescence (IF) labeling (Figure 7) and TEM (Figures 8A & B) support the results of our biochemical assays in demonstrating that HA-o release from NPs with the higher HA-o loadings (5% w/w and 8% w/w) significantly enhances elastic matrix deposition relative to that generated in cultures supplemented with 3% w/w HA-o loaded NPs or blank NPs (IF only) or NP-free cell layers (TEM only). In the TEM, amorphous elastin deposits and microfibrils were identified based on (a) their classic patterns of staining, that has been well described for elastic fiber components [52] (gray staining with phosphotungstic acid for glutaraldehyde and OsO4-sequentially fixed amorphous elastin and darker staining of microfibrils with uranyl acetate and lead citrate), (b) the location of these two differentially stained components (amorphous elastin core and microfibrils ringing the periphery), and (c) consistent agreement in diameters of the observed clusters with that of the cross-sectional diameters of typical mature elastic fibers (1–2 μm). TEM showed the matrix in HA-o NP supplemented cultures to be composed of numerous agglomerating amorphous elastin deposits associated with a network of peripheral network of microfibrils, typical of normal elastic fiber formation (Figure 8B). This is likely due to the elastogenic potential of HA-o, through its association with verisican as well as its role as a highly anionic GAG. Highly anionic GAGs are believed to promote elastic fiber formation by electrostatically binding to the unoxidized lysine residues of newly synthesized tropoelastin during their association with microfibrils [53]. Differently, only a few sporadic, and isolated amorphous elastin deposits were seen in cell layers in NP-untreated controls.
While delivery of HA-o NPs to cell cultures, as we have performed in this study, is relatively straightforward, their in vivo delivery to the AAA wall, which we propose to subsequently pre-clinically assess in a rat model of the disease, is more complicated. Based on the results of our present in vitro studies, for initial attempts at in vivo delivery in rat AAA models, NPs will be loaded with 8% w/w of HA-o, and delivered at a dose that results in a final volumetric density equivalent to 0.5 mg of NPs per cubic cm of AAA tissue, since these are the conditions that we have determined here to maximally deliver HA-o to cells without being cytotoxic. Our future studies will focus on identifying a route of delivery (periaortic versus intraluminal) for NPs that most effectively ensures uptake and localization at the site of intended tissue repair (medial and adventitial layers of the aorta). Based on feedback provided by NP retention studies within AAA tissues, we expect future studies to further improvise on methods of modifying NP-surfaces to more specifically target binding to cell surface proteins that are uniquely expressed within AAA tissues, so as to improve retention within the AAA wall. Intraluminal NP delivery to AAAs may be especially challenging since many AAAs contain an intraluminal thrombus (ILT) [54], which despite being porous, are likely to present a significant barrier to diffusion or convective outflow of NPs from circulation into the aortic wall. In this light, further innovation in NP design will be required to effect controlled thrombolysis to facilitate delivery of nanotherapeutics for pro-matrix regenerative effects.
5. Conclusion
PLGA nanoparticles encapsulated with varying percent loadings of HA-o were synthesized and were shown to exhibit mean hydrodynamic sizes of ~350 nm which is appropriate for exclusion by cells in the extracellular space where elastic matrix assembly occurs. These NPs were formed using 0.25% PVA to impart a negative charge of 30 mV which does not affect cell viability when used at 0.5 mg/mL. The NPs were found to maintain HA-o release over an extended period (>30 days) at steady-state levels. The NPs and released HA-o induced dose dependent increases in elastic matrix synthesis. Furthermore, they significantly enhanced LOX production, and led to improved deposition of a microfibrillar network by the SMCs, which were responsible for the high degree of elastic matrix crosslinking and fiber formation observed. In addition the HA-o NPs did not upregulate MMP synthesis in the EaRASMC cultures, based on western blots. Future studies will focus on targeted delivery of these optimized PLGA HA-o NP formulations to induced AAAs within a rat model.
Supplementary Material
Acknowledgments
The authors acknowledge funding from the NIH (Grant R01HL092051 awarded to Anand Ramamurthi and SOURCE summer funding by the Case Alumni Association (CAA) awarded to Andrew Sylvester.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kothapalli CR, Gacchina CE, Ramamurthi A. Utility of Hyaluronan Oligomers and Transforming Growth Factor-Beta1 Factors for Elastic Matrix Regeneration by Aneurysmal Rat Aortic Smooth Muscle Cells. Tissue Eng Part A. 2009;15:3247–58. doi: 10.1089/ten.tea.2008.0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson SG, Ashton HA, Gao L, Scott RAP. Screening men for abdominal aortic aneurysm: 10 year mortality and cost effectiveness results from the randomised Multicentre Aneurysm Screening Study. Bmj. 2009;338:2307. doi: 10.1136/bmj.b2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Upchurch GR, Jr, Schaub TA. Abdominal aortic aneurysm. Am Fam Physician. 2006;73:1198–204. [PubMed] [Google Scholar]
- 4.Brewster DC, Jones JE, Chung TK, Lamuraglia GM, Kwolek CJ, Watkins MT, et al. Long-term outcomes after endovascular abdominal aortic aneurysm repair: the first decade. Ann Surg. 2006;244:426–38. doi: 10.1097/01.sla.0000234893.88045.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kothapalli CR, Ramamurthi A. Biomimetic Regeneration of Elastin Matrices Using Hyaluronan and Copper Ion Cues. Tissue Eng Part A. 2009;1:103–12. doi: 10.1089/ten.tea.2007.0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joddar B, Ramamurthi A. Fragment size- and dose-specific effects of hyaluronan on matrix synthesis by vascular smooth muscle cells. Biomaterials. 2006;27:2994–3004. doi: 10.1016/j.biomaterials.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 7.Joddar B, Ramamurthi A. Elastogenic effects of exogenous hyaluronan oligosaccharides on vascular smooth muscle cells. Biomaterials. 2006;27:5698–707. doi: 10.1016/j.biomaterials.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 8.Gacchina CE, Deb P, Barth JL, Ramamurthi A. Elastogenic inductability of smooth muscle cells from a rat model of late stage abdominal aortic aneurysms. Tissue Eng Part A. 2011;17:1699–711. doi: 10.1089/ten.tea.2010.0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sivaraman B, Ramamurthi A. Multifunctional nanoparticles for doxycycline delivery towards localized elastic matrix stabilization and regenerative repair. Acta Biomater. 2013;9:6511–25. doi: 10.1016/j.actbio.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guzman LA, Labhasetwar V, Song C, Jang Y, Lincoff AM, Levy R, et al. Local intraluminal infusion of biodegradable polymeric nanoparticles. A novel approach for prolonged drug delivery after balloon angioplasty. Circulation. 1996;94:1441–8. doi: 10.1161/01.cir.94.6.1441. [DOI] [PubMed] [Google Scholar]
- 11.Labhasetwar V, Song C, Humphrey W, Shebuski R, Levy RJ. Arterial uptake of biodegradable nanoparticles: Effect of surface modifications. J Pharm Sci. 1998;87:1229–34. doi: 10.1021/js980021f. [DOI] [PubMed] [Google Scholar]
- 12.Song CX, Labhasetwar V, Murphy H, Qu X, Humphrey WR, Shebuski RJ, et al. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. J Control Release. 1997;43:197–212. [Google Scholar]
- 13.Anidjar S, Salzmann JL, Gentric D, Lagneau P, Camilleri JP, Michel JB. Elastase-induced Experimental Aneurysms in Rats. Circulation. 1990;82:973–81. doi: 10.1161/01.cir.82.3.973. [DOI] [PubMed] [Google Scholar]
- 14.Jacobsen MD, Weil M, Raff MC. Role of Ced-3/ICE-family proteases in staurosporine-induced programmed cell death. J Cell Biol. 1996;133:1041–51. doi: 10.1083/jcb.133.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weil M, Jacobson MD, Coles HS, Davies TJ, Gardner RL, Raff KD, et al. Constitutive expression of the machinery for programmed cell death. J Cell Biol. 1996;133:1053–9. doi: 10.1083/jcb.133.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papadopoulos NG, Dedoussis GVZ, Spanakos G, Gritzapis AD, Baxevanis CN, Papamichail M. An improved fluorescence assay for the determination of lymphocyte-mediated cytotoxicity using flow cytometry. J Immunol Methods. 1994;177:101–11. doi: 10.1016/0022-1759(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 17.Wang XM, Terasaki PI, Rankin GW, Jr, Chia D, Hui Ping Z, Hardy S. A new microcellular cytotoxicity test based on calcein AM release. Hum Immunol. 1993;37:264–70. doi: 10.1016/0198-8859(93)90510-8. [DOI] [PubMed] [Google Scholar]
- 18.Hedberg EL, Shih CK, Solchaga LA, Caplan AI, Mikos AG. Controlled release of hyaluronan oligomers from biodegradable polymeric microparticle carriers. J Control Release. 2004;100:257–66. doi: 10.1016/j.jconrel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 19.Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay procedure. Anal Biochem. 1980;102:344–52. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 20.Gacchina CE, Ramamurthi A. Impact of pre-existing elastic matrix on TGFβ1 and HA oligomer-induced regenerative elastin repair by rat aortic smooth muscle cells. J Tissue Eng Regen Med. 2011;5:85–96. doi: 10.1002/term.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kothapalli CR, Ramamurthi A. Induced elastin regeneration by chronically activated smooth muscle cells for targeted aneurysm repair. Acta Biomat. 2010;6:170–8. doi: 10.1016/j.actbio.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kothapalli CR, Gacchina CE, Ramamurthi A. Utility of Hyaluronan Oligomers and Transforming Growth Factor-Beta1 Factors for Elastic Matrix Regeneration by Aneurysmal Rat Aortic Smooth Muscle Cells. Tissue Eng. 2009;15:3247–60. doi: 10.1089/ten.tea.2008.0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kothapalli CR, Ramamurthi A. Benefits of concurrent delivery of hyaluronan and IGF-1 cues to regeneration of crosslinked elastin matrices by adult rat vascular cells. J Tissue Eng Regen Med. 2008;2:106–16. doi: 10.1002/term.70. [DOI] [PubMed] [Google Scholar]
- 24.Kothapalli CR, Taylor PM, Smolenski RT, Yacoub MH, Ramamurthi A. Transforming Growth Factor Beta 1 and Hyaluronan Oligomers Synergistically Enhance Elastin Matrix Regeneration by Vascular Smooth Muscle Cells. Tissue Eng. 2009;15:501–11. doi: 10.1089/ten.tea.2008.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rekhter MD. Collagen synthesis in atherosclerosis: too much and not enough. Cardiovasc Res. 1999;41:376–84. doi: 10.1016/s0008-6363(98)00321-6. [DOI] [PubMed] [Google Scholar]
- 26.Sivaraman B, Bashur C, Ramamurthi A. Advances in biomimetic regeneration of elastic matrix structures. Drug Deliv Transl Res. 2012;2:323–50. doi: 10.1007/s13346-012-0070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gacchina CE, Deb PP, Barth JL, Ramamurthi A. Elastogenic Inductability of Smooth Muscle Cells from a Rat Model of Late Stage Abdominal Aortic Aneurysms. Tissue Eng. 2011;17:1699–711. doi: 10.1089/ten.tea.2010.0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galeano M, Polito F, Bitto A, Irrera N, Campo GM, Avenoso A, et al. Systemic administration of high-molecular weight hyaluronan stimulates wound healing in genetically diabetic mice. Biochim Biophys Acta. 2011;1812:752–9. doi: 10.1016/j.bbadis.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Sapet C, Laurent N, de Chevigny A, Le Gourrierec L, Bertosio E, Zelphati O, et al. High transfection efficiency of neural stem cells with magnetofection. Biotechniques. 2011;50:187–9. doi: 10.2144/000113628. [DOI] [PubMed] [Google Scholar]
- 30.Al-Ghananeem AM, Malkawi AH, Muammer YM, Balko JM, Black EP, Mourad W, et al. Intratumoral Delivery of Paclitaxel in Solid Tumor from Biodegradable Hyaluronan Nanoparticle Formulations. AAPS PharmSciTech. 2009;10:410–7. doi: 10.1208/s12249-009-9222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int J Nanomedicine. 2011;6:877–95. doi: 10.2147/IJN.S18905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li S. Hydrolytic Degradation Characteristics of Aliphatic Polyesters Derived from Lactic and Glycolic Acids. J Biomed Mater Res. 1999;48:342–53. doi: 10.1002/(sici)1097-4636(1999)48:3<342::aid-jbm20>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 33.Hedberg EL, Shih CK, Solchaga LA, Caplan AI, Mikos AG. Controlled release of hyaluronan oligomers from biodegradable polymeric microparticle carriers. J Control Release. 2004;100:257–66. doi: 10.1016/j.jconrel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 34.Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat Nanotechnol. 2007;2:469–78. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 35.Fahmy TM, Demento SL, Caplan MJ, Mellman I, Saltzman WM. Design opportunities for actively targeted nanoparticle vaccines. Nanomedicine. 2008;3:343–55. doi: 10.2217/17435889.3.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy: Rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47:3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 37.Mohanraj V, Chen Y. Nanoparticles – A Review. Trop J Pharm Res. 2006;5:561–73. [Google Scholar]
- 38.Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–91. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al-Remawi MM. Properties of Chitosan Nanoparticles Formed Using Sulfate Anions as Crosslinking Bridges. Amer J of App Scu. 2012;9:1091. [Google Scholar]
- 40.Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (D, L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82:105–14. doi: 10.1016/s0168-3659(02)00127-x. [DOI] [PubMed] [Google Scholar]
- 41.Kothapalli CR, Taylor PM, Smolenski RT, Yacoub MH, Ramamurthi A. Transforming growth factor beta 1 and hyaluronan oligomers synergistically enhance elastin matrix regeneration by vascular smooth muscle cells. Tiss Engr Prt A. 2009;15:501–11. doi: 10.1089/ten.tea.2008.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14:617–23. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- 43.Joddar B, Ibrahim S, Ramamurthi A. Impact of delivery mode of hyaluronan oligomers on elastogenic responses of adult vascular smooth muscle cells. Biomaterials. 2007;28:3918–27. doi: 10.1016/j.biomaterials.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fraser JR, Laurent TC, Engstrom-Laurent A, Laurent UG. Elimination of hyaluronic acid from the blood stream in the human. Clin Exp Pharmacol Physiol. 1984;11:17–25. doi: 10.1111/j.1440-1681.1984.tb00235.x. [DOI] [PubMed] [Google Scholar]
- 45.Onoda M, Yoshimura K, Aoki H, Ikeda Y, Morikage N, Furutani A, et al. Lysyl oxidase resolves inflammation by reducing monocyte chemoattractant protein-1 in abdominal aortic aneurysm. Atherosclerosis. 2010;208:366–9. doi: 10.1016/j.atherosclerosis.2009.07.036. [DOI] [PubMed] [Google Scholar]
- 46.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–8. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 47.Huffman MD, Curci JA, Moore G, Kerns DB, Starcher BC, Thompson RW. Functional importance of connective tissue repair during the development of experimental abdominal aortic aneurysms. Surgery. 2000;128:429–38. doi: 10.1067/msy.2000.107379. [DOI] [PubMed] [Google Scholar]
- 48.Weissen-Plenz G, Eschert H, Völker W, Sindermann JR, Beissert S, Robenek H, et al. Granulocyte Macrophage Colony-Stimulating Factor Deficiency Affects Vascular Elastin Production and Integrity of Elastic Lamellae. J Vasc Res. 2008;45:103–10. doi: 10.1159/000109819. [DOI] [PubMed] [Google Scholar]
- 49.Kagan HM, Li WD. Lysyl oxidase: Properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88:660–72. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 50.Isnard N, Legeais JM, Renard G, Robert L. Effect of hyaluronan on MMP expression and activation. Cell Biol Int. 2001;25:735–9. doi: 10.1006/cbir.2001.0759. [DOI] [PubMed] [Google Scholar]
- 51.Isnard N, Robert L, Renard G. Effect of sulfated GAGs on the expression and activation of MMP-2 and MMP-9 in corneal and dermal explant cultures. Cell Biol Int. 2003;27:779–84. doi: 10.1016/s1065-6995(03)00167-7. [DOI] [PubMed] [Google Scholar]
- 52.Greenlee TK, Ross R, Hartman JL. Fine structure of elastic fibers. J Cell Biol. 1966;30:59–71. doi: 10.1083/jcb.30.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fornieri C, Baccarani-Contri M, Quaglino D, Jr, Pasquali-Ronchetti I. Lysyl oxidase activity and elastin/glycosaminoglycan interactions in growing chick and rat aortas. J Cell Biol. 1987;105:1463–9. doi: 10.1083/jcb.105.3.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. The Lancet. 2005;365:1577–89. doi: 10.1016/S0140-6736(05)66459-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.