

Abstract
Three-dimensional (3D) bioprinting, a flexible automated on-demand platform for the free-form fabrication of complex living architectures, is a novel approach for the design and engineering of human organs and tissues. Here, we demonstrate the potential of 3D bioprinting for tissue engineering using human skin as a prototypical example. Keratinocytes and fibroblasts were used as constituent cells to represent the epidermis and dermis, and collagen was used to represent the dermal matrix of the skin. Preliminary studies were conducted to optimize printing parameters for maximum cell viability as well as for the optimization of cell densities in the epidermis and dermis to mimic physiologically relevant attributes of human skin. Printed 3D constructs were cultured in submerged media conditions followed by exposure of the epidermal layer to the air–liquid interface to promote maturation and stratification. Histology and immunofluorescence characterization demonstrated that 3D printed skin tissue was morphologically and biologically representative of in vivo human skin tissue. In comparison with traditional methods for skin engineering, 3D bioprinting offers several advantages in terms of shape- and form retention, flexibility, reproducibility, and high culture throughput. It has a broad range of applications in transdermal and topical formulation discovery, dermal toxicity studies, and in designing autologous grafts for wound healing. The proof-of-concept studies presented here can be further extended for enhancing the complexity of the skin model via the incorporation of secondary and adnexal structures or the inclusion of diseased cells to serve as a model for studying the pathophysiology of skin diseases.
Introduction
Skin is the largest organ of the human body, and it plays a vital role in maintaining homeostasis as well as in providing protection from the external environment.1–3 The highly complex, hierarchical, and stratified structure of the skin provides a physical barrier to the entry of xenobiotics into the body while regulating the transport of water and small metabolites out of the body. Wounds, originating from physical or chemical trauma, can significantly compromise the skin barrier and impair its physiological functions. In instances in which a considerable amount of the skin has been lost to injuries, it becomes critical to replace the impaired skin via grafts to protect water loss from the body as well as to mitigate the risk posed by opportunistic pathogens. Skin grafts can also greatly facilitate the wound-healing process and can potentially restore the barrier and regulatory functions at the site of the wound.4–7 Beyond grafts, tissue engineered skin can serve as an extremely valuable in vitro platform to evaluate the permeability as well as the adverse inflammatory responses of topical agents in a high-throughput manner during the preliminary stages of transdermal and topical drug discovery and formulation development.8–12 Engineered skin provides several advantages compared with animal skin by better mimicking human skin physiology as well as by alleviating ethical concerns and conforming to emerging regulations on animal use.13 In addition, engineered skin models can provide fundamental insights into the etiology of skin diseases as well as elucidate the pathophysiological mechanisms in skin disease progression and treatment.14–17
Over the past four decades, several groups in industry and academia have invested significant efforts in the design and engineering of human skin with early efforts largely focused on developing skin grafts for wounds.18–20 These were subsequently followed by studies focused on developing in vitro skin models for the assessment of permeability of drugs and excipients across the skin.21–24 A few studies have also attempted to recreate the immune function of the human skin in addition to its physical barrier properties with reasonable success.25,26 These efforts have collectively led to a broad range of approaches for engineering human skin and a variety of in vitro skin models available for research. The typical approach to engineering skin begins by simplifying its complexity and representing it as a two-compartment tissue. The first of these is the multi-stratified epidermis that is composed of the basal, spinous, and granular layers in the live layer, all of which are represented by keratinocytes (KCs) at varying degrees of differentiation; and the dead stratum corneum is represented by terminally differentiated KCs (corneocytes) in a lipid-rich bilayer matrix. The second compartment, dermis, is typically represented by synthetic substrates (e.g., nylon and polycarbonate) or acellular matrix protein scaffolds (e.g., collagen, glycosaminoglycans, and fibrin) or dead de-epidermized dermis or fibroblasts (FBs) that are dispersed within protein scaffolds. Skin tissue engineering typically involves isolating KCs from full-thickness or split-thickness skin by enzymatic digestion, and growing them at the air–liquid interface (ALI) on a dermal surrogate. KCs receive nutrients from the bottom surface of the culture while being pushed upward in a process of progressive differentiation over 3–4 weeks within the culture. This approach results in a skin tissue that grossly mimics the morphological and biological features of the human skin.
Although conventional tissue engineering strategies have facilitated the design and development of the first few generations of skin grafts and models, there remains much room for improvement in skin tissue engineering. Much effort to date has focused on the incorporation of only two types of cells, that is, KCs and FBs, in the epidermis and dermis, respectively, despite the skin being a repository of close to a dozen lineage-differentiated cells as well as multipotent stem cells. While this is sufficient to reproduce dominant structures within the skin, it fails to capture subtle but critical cell–cell interactions, signaling events, and, quite importantly, adaptive immune functions of the skin. Currently, a key limiting factor in incorporating immune cells in the skin is the challenge of maintaining the cells in their naïve immune state, and it is likely related to their inefficient incorporation within the specific skin strata in addition to the absence of other functionally important cells. Contemporary approaches to skin engineering do not take into account the precise positioning of the cells within the individual strata.27–29 This, however, is critical for faithfully reproducing the cell–cell and cell–matrix interactions within the skin. Furthermore, these approaches require incubation periods of 3–4 weeks or greater to form fully differentiated skin. The throughput available from these models is relatively low and, thus, a limiting factor for conducting high-throughput studies.
Several technologies have recently become available for tissue engineering that adopt a fundamentally different and novel approach.30–40 Among these, three-dimensional (3D) free-form fabrication, generally described as 3D bioprinting, offers significant advantages compared with conventional skin tissue engineering. Capable of dispensing live cells, soluble factors, and phase-changing hydrogels in a desired pattern while maintaining very high cell viability,41–45 this approach has tremendous potential in the fabrication of 3D skin tissue. By precisely locating multiple types of matrix materials and cells in a layer-by-layer assembly, various functional tissues can be fabricated with appropriate structures and cell compositions in a wide range of sizes, in a high throughput, and in a highly reproducible fashion.41–45 The multi-layered and highly stratified structure of the skin makes it a perfect example to demonstrate the strengths and advantages of the 3D bioprinting approach while overcoming some of the limitations of traditional tissue engineering schemes. Here, we describe our proof-of-concept studies on the use of 3D bioprinting to engineer human skin in a layer-by-layer assembly process using FBs and KCs as representative cell types. We present our results on optimization studies along with histological and immunofluorescence characterization of the printed 3D human skin. The work presented here sets the stage for further development and engineering of human skin that incorporates multiple skin cells, and adnexal as well as secondary structures.
Materials and Methods
Development of a 3D bioprinter
To afford the flexibility of controlling the geometrical micro- and macro-cellular environments and the cell-to-cell interactions in 3D, we have developed a robotic bioprinting platform based on the 3D solid freeform fabrication technology. This platform has been used and reported in previous studies.41 It consists of eight independently controlled cell-dispensing channels that can precisely place cells, extracellular matrix (ECM), scaffold materials, and growth factors in any user-defined 3D pattern. Each dispenser is independently operated by electromechanical valves and mounted on a three-axis, high-precision, and xyz robotic stage. The liquid materials are dispensed by pneumatic pressure during the gate-opening phase of the micro-valves. In addition, the volume of dispensed droplets (i.e., drop size) can be manipulated by controlling valve opening time and air pressure. The noncontact dispensers are capable of dispensing cells in volumes as low as 15 nL with high precision and high viability, and they are compatible with a variety of hydrogels that are used in tissue engineering. The dispense volume can be adjusted by digitally controlling the pressure and pulse duration (100∼800 μs). Normal operation allows for continuous dispensing with an actuation frequency of 1 kHz, providing high throughput printing capability. The minimum resolution of the printing varies with the viscosity of the material. For aqueous materials (e.g., water and cell culture media), the minimum resolution is ∼100 μm, and it is higher for viscous materials (e.g., collagen and matrix proteins). The resolution can be varied systematically by controlling the volume of the dispensed droplets. A user-friendly software interface facilitates on-demand generation of spatial patterns of the materials in 3D at sub-cellular accuracy.
Cell culture and hydrogel preparation
FBs (HFF-1, from ATCC) and KCs (HaCaT, gift from Dr. Torsten Wittmann at University of California, San Francisco) were used to construct the 3D skin tissue in this study. Both cell lines were cultured at 37°C in 5% CO2 in Dulbecco's modification of Eagle's Medium (ATCC) supplemented with 15% fetal bovine serum (Thermo Scientific) and 1% Penicillin/Streptomycin (Mediatech). Culture media was changed every 3 days. The cells were routinely passaged in tissue culture flasks, and they were discarded after 20 passages to ensure representation of key characteristics. Before printing, FBs and KCs were harvested using 0.25% Trypsin/ethylenediaminetetraaceticacid (Mediatech) at a desired suspension density. They were then maintained as cell suspensions on ice until they were ready to be loaded into the syringes for printing.
Collagen hydrogel precursor (Rat tail, type I; BD Biosciences) was used as a scaffold material for printing. The collagen precursor from stock was diluted to 3.0 mg/mL with 1× Dulbecco's phosphate-buffered saline (without calcium and magnesium) and was maintained on ice until it was ready to be loaded into a syringe (which serves as a printer cartridge) for printing. Printing pressure and dispenser valve opening times (pulse duration) are crucial parameters for the proper printing of various biomaterials, hydrogels, and cells. These parameters were determined based on the viscosity of the biomaterials being dispensed. The lowest pressure that showed stable dispensing without clogging was used for the printing of collagen precursors and cell suspensions. Since collagen precursor is more viscous than cell suspensions, higher pressure was employed for the printing of collagen compared with cells. Pressure values in the range of 2.5–2.7 psi and 750 μs valve opening times were used for collagen printing, leading to droplet volume of ∼52 nL. For cell printing, ∼28 nL droplets were obtained by using 1.4–1.5 psi of pressure and 750 μs of valve opening time (Table 1).
Table 1.
Summary of Printing Parameters Used in This Study
| Printing parameters | Collagen | Cell suspension |
|---|---|---|
| Air pressure | 2.5–2.7 psi | 1.4–1.5 psi |
| Valve opening time (pulse duration) | 750 μs | 750 μs |
| Droplet volume | 52.77±3.81 nL | 28.53±3.15 nL |
| Droplet spacing (resolution) | 500 μm | 500 μm |
| Pattern size | 6×6 mm | 4×4 mm |
| Concentration/density | 3.0 mg/mL | 0.5–5 million cells/mL |
Optimization of cell density and droplet spacing for cell printing
Before the 3D multi-layered printing and co-culturing, we tested different resolutions or droplet spacings (i.e., distance between individual printed droplets) and cell suspension densities to find the optimum conditions for cell viability and proliferation of each cell type. Combinations of various spacings (100, 150, 200, 300, 400, 500, and 750 μm) and cell suspension densities (0.5, 0.75, 1, and 2 million cells/mL for FBs; 0.5, 1, 2, 3, and 5 million cells/mL for KCs) were tested. The dimension of each collagen layer was maintained at 6×6 mm, whereas the dimension for each cell layer was fixed at 4×4 mm (Fig. 1, Top View). A layer of collagen was printed on a petri dish followed by FB or KC cell suspensions on top of the collagen at the desired density and droplet spacing (Fig. 1). The printed collagen-cell constructs were then placed in an incubator for one hour, and culture media was gently added. After 7 days of culture, with media changes on days 3 and 6, cell viability was measured via confocal microscopy using Hoechst 33342 and propidium iodide staining.
FIG. 1.

Printing scheme employed for the generation of single-cell cultures. Two layers of collagen were printed (i, ii) followed by the printing of a one-cell layer (iii). The dimensions of collagen and cell layers were maintained at 6×6 mm and 4×4 mm, respectively. FB, fibroblasts; KC, kerotinocytes. Color images available online at www.liebertpub.com/tec
Construction of 3D skin tissue
The optimum parameters determined from the previously described studies on the printing of individual cell types as monolayers on collagen were used for the construction of 3D skin structures. Construction was achieved using a layer-by-layer fabrication approach as described in Figure 2a. The multi-layered collagen structure with embedded cells was constructed on poly-d-lysine-coated glass-bottom petri dishes (MatTek). Before printing was initiated, nebulized sodium bicarbonate (NaHCO3) vapor was applied onto the petri dish surface, enabling the quick gelation of the first printed collagen layer and thereby increasing its adhesion to the bottom surface. Subsequent to the printing of the first collagen layer, nebulized NaHCO3 vapor was applied onto the printed collagen layer for gelation. To provide a firm base for the next layer of printing, a time lapse of one minute was allowed to facilitate the phase transition of collagen to a gel. In our design scheme, the printed skin structure contains eight collagen layers. These include six collagen layers alternating with three layers of FB layers and two collagen layers separating the stacked FB layers from KCs as shown in Figure 2b. Two KC layers were printed to achieve the desired cell density within the epidermis. On completion of the printing steps, the composite structure was placed in an incubator (37°C, 5% CO2) for 1 h to complete the collagen gelation. The whole tissue structure was then submerged in the culture media. Media was changed once in every 3 days.
FIG. 2.

Construction of three-dimensional (3D) skin tissue. (a) Layer-by-layer printing of collagen matrix, KCs, and FBs to construct the dermal and epidermal compartments in a single structure. (b) Schematic of the 3D printed skin tissue showing the cross-section (left) and top view (right). Color images available online at www.liebertpub.com/tec
ALI culture
Once the individual steps for printing 3D multi-layered cultures in a petri dish were established, a similar scheme was used to print the skin structures on a 12-well transwell membrane support (pore size: 0.4 μm; Corning). Transwell supports facilitate the printed cultures to be grown at the ALI, a condition in which KCs are cultured at the air–liquid boundary to promote progressive terminal differentiation into corneocytes and the formation of the stratum corneum. This step is essential for the formation of a stratified multi-layered tissue similar to the skin.46 The dimensions of the printed structure, number of printed layers, and the printing parameters (pressure, valve opening time, droplet spacing, and cell suspension density) on transwell membrane inserts were identical to those optimized for printing on petri dish supports. On completion of the printing sequence, the skin constructs were placed in an incubator (37°C, 5% CO2) for one hour to facilitate complete collagen phase transition to a gel. Media was changed every 3 days. The printed structures were cultured for 4–8 days under submerged conditions followed by exposing them to the ALI by adjusting the media levels in the inserts. This step causes the KCs to proliferate and completely cover the top of the collagen surface. To increase the extracellular matrix production and to facilitate the maturation of the tissue construct, the following components were added to the culture media: 1× Human Keratinocyte Growth Supplement (Life Technology; 0.2 ng/mL human epidermal growth factor, 0.18 μg/mL hydrocortisone, 5 μg/mL bovine transferrin, 5 μg/mL bovine insulin, and 0.2% v/v bovine pituitary extract), 50 μg/mL l-ascorbic acid, and 1.3 mM calcium chloride. Media was changed every day during the culture at the ALI for approximately 14 days.
Conventional 3D skin constructs
Conventional 3D skin constructs were created using the manual deposition method.28 Final concentrations of collagen, cell volumetric density of FBs, and surface area density of KCs were calculated from printing parameters (pattern size, droplet spacing, droplet volume, and total number of printed layers; noted in Table 1) and were used to create the conventional 3D skin constructs. Briefly, dermal equivalents, comprising FBs (final cell density 2.3×105 cells/mL) mixed with collagen matrix (final concentration 2.66 mg/mL) in 500 μL volume, were grown on 12-well plate transwell membranes. The dermal structure was cultured until it fully contracted, forming a concave area over 7 days (Fig. 6). The media was then removed, and then, 500 μL of fresh media containing KCs (surface seeding density 1.4×105 cells/cm2) was placed gently on the top of the dermal layer for 3 h. It was then followed by the addition of fresh media to fully submerge the skin culture. One week after KC seeding (Day 14), the cultures were allowed to grow at the ALI for 7 days.
FIG. 6.

Shape and form of printed skin tissue. A comparison of skin tissues fabricated via 3D bioprinting and manual deposition indicates that printed skin samples (a, b) retain their form (dimensions) and shape, whereas manually deposited structures (c, d) shrink and form concave shapes (buckle) under submerged culture condition after 7 days. Color images available online at www.liebertpub.com/tec
Live/dead staining of printed tissue
The printed skin constructs were cultured for 7 days with media changes every 3 days. At the end of the culture period, old media was replaced with fresh media containing two nuclei stains, Hoechst 33342 and propidium iodide, and incubated for 8 h in the incubator. Hoechst 33342 was used at a concentration of 10 μg/mL, whereas propidium iodide was used at a concentration of 2 μg/mL. Hoechst 33342 stains all cell nuclei blue, while propidium iodide stains only dead cell nuclei red. After incubation, the samples were washed with fresh media, thrice for 10 min each. They were then imaged using a confocal microscope using two lasers: 405 nm laser for exciting the Hoechst dye and 543 nm laser for exciting the propidium iodide dye. The images were collected as Z-stacks and then compressed onto a single plane using Zeiss LSM software. The images were transferred to Image J software (NIH) to calculate live and dead cells. The ratio of viable cells to the total number of cells is expressed as percentage viability.
Thickness measurement of printed skin tissues
Phase-contrast microscopy imaging was employed to determine the thickness of skin constructs. The focal planes of the transwell membrane and uppermost KCs were identified under a bright-field microscope using a 10×objective, and the z-axis positions of the microscope at the corresponding focal planes were recorded. The difference between two z-positions, that is, the distance between the transwell membrane and the uppermost KCs, was noted as the thickness of the sample. Thickness of the skin cultures was measured every 1–3 days from 12 replicates during the submerged culture and ALI culture conditions. The measurements were performed on live samples without additional staining or fixing processes.
Histology and immunofluorescence
After the culture of skin samples for 7 and 14 days at the ALI, the samples were fixed with 4% paraformaldehyde. They were then embedded in paraffin and sectioned using a microtome into 5 μm-thick slices. The slices were stained with hemotoxylin and eosin (H&E) using standard protocols.28 The presence of N-cadherin was determined using Mouse Anti-Human N-cadherin antibody (4 μg/mL; Millipore), followed by incubation with the Alexa Fluor 594 Goat Anti-Mouse IgG (H+L) antibody (4 μg/mL; Invitrogen) to evaluate its phenotypical and morphological resemblance with that of human skin. Each antibody incubation step was followed by three washing steps with phosphate-buffered saline, for 10 min each.
Results
Optimization of printing parameters for monolayer cultures
Various hydrogel materials can be employed to construct 3D tissue structures. In this particular study, collagen gel (type I, rat tail) was used as the primary matrix to construct the skin tissue. Typically, gelation of matrix proteins is achieved by modulating the pH or temperature or both. Although this approach is well suited for thin structures, it can lead to heterogeneous regions of gelled and nongelled matrices due to diffusion or heat transfer limitations in thick structures (1–3 mm). Large gradients in pH or temperature may also result in undesired effects on the cells. To avoid these issues, we have utilized an approach that relies on printing cells and proteins encapsulated in culture medium and using aerosolized or nebulized NaHCO3 as a cross-linker for collagen. The culture medium within the cell and protein droplets provides a buffer condition that protects cells from potential damage, while NaHCO3 vapors provide a gentle but effective approach to achieve gelation. The use of gentle pressure (1.4–1.5 psi) for cell printing also aids in maintaining cell function. In our control experiments, cells printed directly in two dimensions on tissue culture plates presented identical morphology, proliferation, and ability to form tight junctions compared with the cells plated manually.
In skin tissue engineering, achieving representative cell density within each layer (epidermis and dermis) is critical to obtaining a functionally and morphologically representative tissue. We control the cell density within each layer by controlling two key printing parameters: cell suspension density in each droplet (cells/mL) and the spacing between individual droplets (μm) within the 3D matrix. To optimize these printing parameters, we first tested various combinations of cell suspension densities and droplet spacings in individual layers. Overall, as expected, cell density within the matrix varied proportionally with cell suspension density and inversely with droplet spacing for both KCs and FBs. Irrespective of the specific combination of density and spacing, we observed very high cell viability for both KCs and FBs. Figure 3 shows results of the viability assay at select test conditions for FBs (Fig. 3a–f, m) and KCs (Fig. 3g–l, n). The viability of FBs was generally ∼98% except at very low cell suspension density (0.5–0.75 million cells/mL) and large droplet spacing (400–750 μm) as seen from Figure 3a and b (84–89% viability). This is likely due to the lack of cell–cell communication at relatively low surface coverage. Similar to FBs, the viability of KCs was also typically very high, ∼98%, except at very high cell suspension densities (2–5 million cells/mL) and small droplet spacing (200–400 μm) as seen in Figure 3j and l. At very large droplet spacings of ∼750 μm or greater, KCs preferred to grow as individual islands in contrast to uniform monolayers, albeit still at very high viability (Fig. 3g; 94% viability).
FIG. 3.

Optimization of printing parameters for individual cell cultures. Cell suspension density and resolution (spacing between droplets) were varied over a broad range for FBs (a–f) and keratinocytes (KCs) (g–l) printed in monolayers. Cell viability was assessed at each condition after 7 days in culture (m, n). Color images available online at www.liebertpub.com/tec
After our observation that cell viability is very high over a very broad range of cell suspension densities and droplet spacing, the two parameters were adjusted to reflect the cell surface density (cells/cm2) of KCs in the epidermis and the cell volumetric density (cells/mL) of FBs in the dermis. To represent the sparse cell distribution within the dermis and dense cell distribution within the epidermis, 2 million FBs/mL and 5 million KCs/mL at 500 μm droplet spacing each were chosen as the printing parameters for subsequent steps of this study. The number of collagen layers for the dermis and epidermis was determined based on the thickness of a single collagen layer (∼140 μm) obtained under the printing conditions described in Table 1. A total six collagen layers for the dermis and two collagen layers for the epidermis were printed to obtain the desired dermal and epidermal thicknesses (Fig. 2).
Printing of 3D human skin
Figure 2a illustrates the steps used for the printing of the composite in vitro 3D skin tissue. Printing a single skin tissue sample of the dimensions described in this study takes ∼40 min. However, the printer is capable of printing four samples simultaneously, which requires ∼60 min. This time can, however, be significantly reduced using a multiplexed system with multiple print heads. After the printing process, the skin tissue was cultured in media under submerged conditions and imaged by confocal microscopy. Figure 4a shows the densely distributed KCs in the epidermis and sparsely distributed FBs in the dermis. The FB nuclei are larger and elongated (Fig. 4c), whereas those of KCs are flat and circular in shape (Fig. 4b), as expected. The FBs were found to be sparsely but uniformly distributed in the collagen matrix, as expected, based on the suspension density and droplet spacing. The height of the printed skin structure varied from 1100 to 1400 μm with a majority of the thickness attributed to the dermis containing the FBs (Fig. 5). FBs proliferated within the dermal layer and maintained a sparse distribution. KCs proliferated more rapidly on the top of the collagen matrix and fully covered the surface in 4–7 days. No cell invasion across the dermal and epidermal layers was observed. Rapidly proliferating KCs occasionally expanded beyond the edge of the printed tissue; however, the majority of proliferating cells stayed embedded within the collagen matrix. Three densely packed cell layers of KCs (∼30 μm) were observed on the surface of the printed skin with no detectable difference in the size of their nuclei (data not shown). We found that the printed skin tissue model retained its shape and dimensions without shrinking throughout the culture period. In contrast, conventional 3D skin constructs created by manual deposition exhibited continued shrinking starting at day 2 and were unable to retain their shape or form. Figure 6 depicts the change in the size and shape of the printed skin and the manually deposited skin samples after 7 days of submerged culture.
FIG. 4.

Confocal microscopy imaging of cultured skin. (a) 3D reconstruction of confocal microscopy images of printed skin after 7 days in submerged culture conditions. Live cell nuclei are stained green and present compact and rounded (KC) or large and elongated (FB) morphologies. (b, c) Compressed z-projections of the epidermis and dermis showing KCs and FBs. Color images available online at www.liebertpub.com/tec
FIG. 5.

Thickness of printed skin tissues. Tissue thickness varied significantly over the course of the culture period (3 weeks). A dramatic reduction (2 to 6-fold) in skin thickness was observed at the transition from submerged culture conditions to culture at the air–liquid interface.
ALI culture
Printed skin constructs were grown at the ALI for 7–14 days after 7 days of culture under submerged conditions. During this period, the thickness of the skin constructs reduced dramatically to ∼500 μm within the first 24–48 h and continued to decrease gradually over the culture period to ∼150–200 μm by the end of 10 days (Fig. 5). No substantial reduction in skin thickness was observed on further culture for approximately 14 days at the ALI. We also observed that the skin tissue became progressively more translucent over the ALI culture period, indicating the formation of the stratum corneum by corneocytes formed by terminal differentiation of KCs (Fig. 6a, b).
Structure of printed skin tissue
H&E staining was performed to visualize the structural components of the tissue and to measure the number of nuclei in both the dermal and epidermal layers. The sections at day 7 and 14 of ALI were stained with H&E and are shown in Figure 7 after ALI exposure and culture. As seen in the figure, the epidermal layer is very dense, whereas the dermal compartment is sparsely populated. The number of nuclei-containing layers in the epidermis increased from 3 to 7 from day 7 to 14, indicating the formation of stratified layers on ALI culture. The sections from the printed skin samples were also stained for N-cadherin antibody to visualize the presence of the tight junctions, a unique feature of the epidermal layers. Figure 8 demonstrates the staining pattern of N-cadherin junction at day 14 after ALI exposure. As can be seen from the figure, the punctate pattern in the epidermal compartment is highly visible and is present only in the epidermal layer, indicating the formation of junctions between KCs cell layers.
FIG. 7.

Histology of skin tissue. Printed skin cultures were characterized using hematoxylin and eosin and nuclear staining at day 7 (a, b) and day 14 (c, d) after air–liquid interface (ALI) culture. The epidermal layer is very dense and exhibits compaction with time. The dermis is sparsely populated and shows significant shrinking and compaction with time. Color images available online at www.liebertpub.com/tec
FIG. 8.
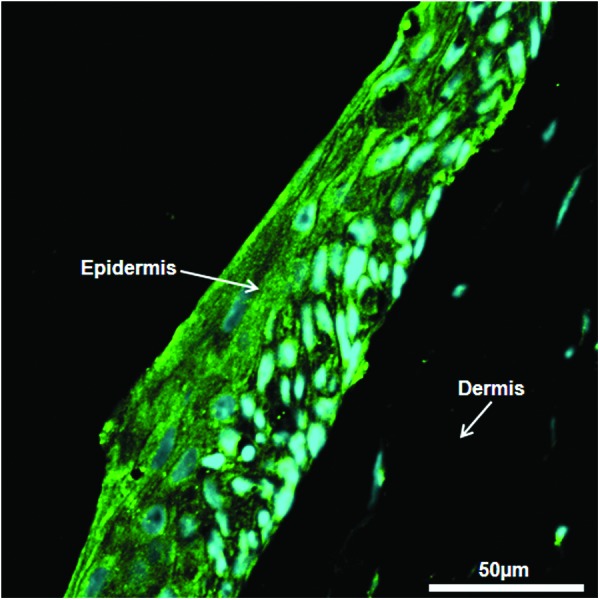
Immunofluorescence of skin tissue. Printed skin structures were stained for N-cadherin tight junctions at day 14 of ALI culture. N-cadherin (green) was observed bordering adjacent epidermal cells (nuclei stained in blue), but it was not detected in the dermal compartment. Color images available online at www.liebertpub.com/tec
Discussion
In this work, we describe the development, optimization, and application of a 3D on-demand cell and protein printing platform for the engineering of biological tissues and organs using human skin as a prototypical example. A multi-layered cell and matrix structure was constructed in which human KCs were grown at the ALI on collagen matrices embedded with human dermal FBs. We were able to effectively control the number of layers, the cell density, and their precise location in the generation of this skin model to reproduce key morphological and biological features of in vivo human skin.
Our studies indicate that printing cells and proteins in nano-to-microliter droplets on planar surfaces similar to ink-jet printing had a minimal effect on cell viability and function either in monocultures or in co-cultures. Viability for both cell types employed in this study, KCs and FBs, was sufficiently high (∼95% or greater for a majority of conditions) with the ability to achieve uniform distribution of FBs in the dermal compartment and that of KCs in the epidermal compartment. Key printing parameters, including cell suspension density and spatial resolution, were, therefore, optimized to specifically reproduce the cellular organization and distribution observed in in vivo human skin. A two-step culture beginning with submerged conditions for 7 days and subsequent exposure to the ALI for 10–14 days was sufficient to effectively generate the three distinct layers of skin-viable dermis and epidermis, and the terminally differentiated stratum corneum, albeit incomplete as noted later. The fully matured skin tissue exhibited 3–7 distinct cell layers in the epidermis, suggesting the stratification of the epidermis into its sub-strata. The presence of tight junctions between KCs in the epidermis indicated a well-formed barrier with extensive cell–cell contacts, as would be expected in vivo.
Histological characterization of mature cultures indicated that a multi-layered epithelium growing on a dermal equivalent containing FBs and collagen matrix is formed by the end of ALI culture. The cellular density, morphology, and thickness are comparable to the tissue constructed by conventional manual deposition techniques28 and to in vivo human skin.47 However, the cell density of FBs in dermis was slightly lower, and the ordered stratification and keratinization, key characteristics of the native epidermis, are incomplete. This is likely due to the several important factors that are related to the specific cells used in this study: (1) The choice of cell types is critical for the stratification and keratinization. To enable proper stratification and keratinization, freshly isolated primary KCs that are unfractionated and uncultured are often used.28 The KCs employed in this study, however, are immortalized and have been passaged in culture for a long time. This can putatively lead to the gradual loss of cell phenotype and function. We anticipate that the incorporation of primary cells or a KC stem cell-enriched population can potentially lead to distinct and enhanced cytokeratin expression with a higher level of tight junction expression in the epidermal compartment. (2) Studies have shown that the epithelial–mesenchymal crosstalk controls epidermal regeneration, implying that the functional status of both the KCs and FBs contributes to epidermal differentiation. This is also influenced by culture conditions. For example, the dermal cells, stimulated by interleukin-1, are able to secrete KC growth factors (such as FGF7), which subsequently stimulate KC proliferation and the development of barrier function in the stratum corneum.48,49 Therefore, we expect that the stratification and keratinization can be further improved with enhanced or chemically defined media and culture conditions. (3) In this model, the complexity of the in vivo situation is not completely replicated. The highly simplified culture models do not display the large dermal cellular heterogeneity and the networks of lymphatic and blood vessels that are physiologically relevant and present in vivo. These can further influence the qualitative and quantitative end points and attributes.
A key advantage of the 3D printing approach is exemplified in the results presented in Figure 6. The 3D-printed skin model maintains its overall shape, structure, and physical dimensions throughout the culture period in contrast to the manually fabricated tissue that shows dramatic shrinkage from day 2 and onward. This result implies that the 3D printing technique can provide better dimensional control of the engineered skin tissue. In constructing the skin grafts, either by printing or by using the conventional manual deposition technique, we employed the exact same matrix materials, at identical concentrations with identical cross-linking reagents and gelation temperatures. In addition, we observed only subtle differences in mechanical properties (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tec) and microstructures of the collagen matrices (Supplementary Fig. S2) created using the two approaches. We, therefore, speculate that the different gelation processes employed in these two methods may lead to subtle differences in the architecture of the collagen gels, which subsequently influence the FB phenotypes and the contraction caused by FBs in the matrix. In comparison to the gelation technique used for constructing conventional grafts that employ the mixing of collagen precursor and cross-linking reagent in bulk, we hypothesize that the layer-by-layer printing of collagen precursor and the application of the cross-linking reagent (NaHCO3) in the dispersed vapor phase may provide more uniform gelling conditions which can prevent the formation of heterogeneous regions of gelled and nongelled matrices. It may also allow the collagen structure to more evenly disperse the contractile forces of FBs to minimize the tissue deformation. During would healing, the differentiation of FBs toward myofibroblasts and the resulting contraction is a major factor in scar formation.50 It is also a clinical challenge when skin grafts are employed to replace skin lost to burns or to injury. It will, therefore, be interesting to investigate whether and how printed skin grafts can potentially reduce tissue contraction during wound healing by examining the status of FB differentiation and the micro-architecture of the printed collagen matrix.
The 3D-engineered skin offers several additional advantages that make it a powerful tool to investigate skin morphogenesis and to understand epidermal biology for (1) enhancing the formation of a well-defined, stratified, and differentiated 3D architecture of the reconstructed epidermis; (2) the key steps to the establishment of a dermal-epithelial cross-talk; (3) the possibility of adding exogenous molecules, enabling the screening of active components regulating skin regeneration; and (4) the feasibility of generating reconstructed skin from any species and genetic background, including those from knockout mice. Compared with manual deposition, the bioprinting method offers the ability to precisely control the cell localization and the size of the construct. It is also relatively simple, which enables the construction of the multi-layered structure in one step instead of two steps at different times as is typical with the manual deposition approach. It can also enable the integration of multiple synthetic or natural scaffold materials and additional cell types (e.g., Langerhans cells, melanocytes, vascular endothelial cells, etc.) at precise locations into the skin model presented in this study. This will enable the design of a skin tissue with more complex structures and functions in the future. The specific bioprinting platform described in these studies can be easily scaled up to include multiplexed dispensers, either to incorporate additional components or to increase throughput. This can facilitate the printing of large skin grafts in a high-throughput and reproducible manner, which is critical for the translation of this platform for medical and/or clinical use. Finally, an important consideration in bioprinting is the stability, activity and retention of the proteins and biomolecules in the printed tissue. These depend on the type of biomaterial, gelation process, gelation temperature, and other cofactors such as salts, buffers, and pH used during printing. Although outside the scope of the present study, we have demonstrated that bioprinted vascular endothelial growth factor-releasing fibrin gel is able to retain the growth factor in its active form for more than 3 days, and, therefore, has the potential to be used as a biomaterial in the printing process to increase the retention and stability of growth factors.43 Similarly, it has been shown that alginate and polyethylene glycol (PEG) hydrogels can be modified to deliver bioactive molecules. These materials can be easily adapted to the printing process to construct 3D cell-gel structures, using a variety of different gelation mechanisms (e.g., CaCl2 or UV light).
Conclusions
We have presented our preliminary studies on fabricating human skin using the 3D bioprinting technology. Further optimization of this platform and our approach can potentially facilitate the fabrication of a more complex human skin model that incorporates secondary and adnexal structures. Such models can lead to significant advancements in our understanding of the human skin as an organ, enabling the engineering of superior wound grafts as well as topical and transdermal formulation development tools while reducing the reliance on animal models. The approach described here can potentially also be translated to the design of disease models for autoimmune diseases of the skin such as psoriasis, atopic dermatitis, allergic contact dermatitis, and vitiligo and models of skin malignancies such as melanoma, leading to the discovery and development of effective therapeutics against these diseases.
Supplementary Material
Disclosure Statement
No competing financial interests exist.
References
- 1.Bos J.D., and Meinardi M.H.The 500 dalton rule for the skin penetration of chemical compounds and drugs. Exp Dermatol 9, 165, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Bouwstra J.A.The skin, a well organized membrane. Colloids Surf A 123, 403, 1997 [Google Scholar]
- 3.Karande P., and Mitragotri S.Transcutaneous immunization: an overview of advantages, disease targets, vaccines, and delivery technologies. In: Prausnitz J.M., Doherty M.F., and Segalman M.A., eds. Ann Rev Chem Biomol Eng 1, 175, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Adams D.C., and Ramsey M.L.Grafts in dermatologic surgery: review and update on full- and split-thickness skin grafts, free cartilage grafts, and composite grafts. Dermatol Surg 31, 1055, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Hierner R., Degreef H., Vranckx J.J., Garmyn M., Massage P., and van Brussel M.Skin grafting and wound healing - the “dermato-plastic team approach”. Clin Dermatol 23, 343, 2005 [DOI] [PubMed] [Google Scholar]
- 6.MacFarlane D.F.Current techniques in skin grafting. Adv Dermatol 22, 125, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Orgill D.P.Excision and skin grafting of thermal burns. N Engl J Med 360, 893, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Karande P., Arora A., Pham T.K., Stevens D., Wojicki A., and Mitragotri S.Transcutaneous immunization using common chemicals. J Control Release 138, 134, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Karande P., Jain A., Arora A., Ho M.J., and Mitragotri S.Synergistic effects of chemical enhancers on skin permeability: a case study of sodium lauroylsarcosinate and sorbitan monolaurate. Eur J Pharmaceut Sci 31, 1, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Karande P., Jain A., Ergun K., Kispersky V., and Mitragotri S.Design principles of chemical penetration enhancers for transdermal drug delivery. Proc Natl Acad Sci U S A 102, 4688, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karande P., Jain A., and Mitragotri S.Discovery of transdermal penetration enhancers by high-throughput screening. Nat Biotechnol 22, 192, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Karande P., and Mitragotri S.High throughput screening of transdermal formulations. Pharmaceut Res 19, 655, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Adler S., Basketter D., Creton S., Pelkonen O., van Benthem J., Zuang V., Andersen K.E., Angers-Loustau A., Aptula A., Bal-Price A., Benfenati E., Bernauer U., Bessems J., Bois F.Y., Boobis A., Brandon E., Bremer S., Broschard T., Casati S., Coecke S., Corvi R., Cronin M., Daston G., Dekant W., Felter S., Grignard E., Gundert-Remy U., Heinonen T., Kimber I., Kleinjans J., Komulainen H., Kreiling R., Kreysa J., Leite S.B., Loizou G., Maxwell G., Mazzatorta P., Munn S., Pfuhler S., Phrakonkham P., Piersma A., Poth A., Prieto P., Repetto G., Rogiers V., Schoeters G., Schwarz M., Serafimova R., Tahti H., Testai E., van Delft J., van Loveren H., Vinken M., Worth A., and Zaldivar J.-M.Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Arch Toxicol 85, 367, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Ali N., Chu C.C., Karagiannis P., Di Meglio P., Skowera A., Napolitano L., Barinaga G., Grys K., Sharif-Paghaleh E., Karagiannis S.N., Peakman M., Lombardi G., and Nestle F.O.A novel skin-resident vitamin D-3-inducible human dendritic cell playing a critical role in tissue homeostasis and immunoregulation, as demonstrated in humanized mouse models of disease. Br J Dermatol 166, e19, 2012 [Google Scholar]
- 15.Aloor H., Peredo C., Hofland H., Smith S.H., Therrien J., and Cote-Sierra J.A new model for evaluating topical treatments for th17 mediated diseases using ex vivo human skin. J Invest Dermatol 133, S3, 2013 [Google Scholar]
- 16.Kuechler S., Henkes D., Eckl K.-M., Ackermann K., Plendl J., Korting H.-C., Hennies H.-C., and Schaefer-Korting M.Hallmarks of atopic skin mimicked in vitro by means of a skin disease model based on flg knock-down. Altern Lab Anim 39, 471, 2011 [DOI] [PubMed] [Google Scholar]
- 17.Semlin L., Schaefer-Korting M., Borelli C., and Korting H.C.In vitro models for human skin disease. Drug Discov Today 16, 132, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Shevchenko R.V., James S.L., and James S.E.A review of tissue-engineered skin bioconstructs available for skin reconstruction. J R Soc Interface 7, 229, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood F.M., Kolybaba M.L., and Allen P.The use of cultured epithelial autograft in the treatment of major burn wounds: eleven years of clinical experience. Burns 32, 538, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Macneil S.Progress and opportunities for tissue-engineered skin. Nature 445, 874, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Morikawa N., Kitagawa T., and Tomihata K.Assessment of the in vitro skin irritation of chemicals using the vitrolife-skin™ human skin model. AATEX 14, 417, 2007 [Google Scholar]
- 22.Netzaff F., Lehr C.M., Wertz P.W., and Schaefer U.F.The human epidermis models episkin (r), skinethic (r) and epiderm (r): an evaluation of morphology and their suitability for testing phototoxicity, irritancy, corrosivity, and substance transport. Eur J Pharmaceut Biopharmaceut 60, 167, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Netzlaff F., Kaca M., Bock U., Haltner-Ukomadu E., Meiers P., Lehr C.M., and Schaefer U.F.Permeability of the reconstructed human epidermis model episkin (r) in comparison to various human skin preparations. Eur J Pharmaceut Biopharmaceut 66, 127, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Schafer-Korting M., Bock U., Diembeck W., Duesing H.J., Gamer A., Haltner-Ukomadu E., Hoffmann C., Kaca M., Kamp H., Kersen S., Kietzmann M., Korting H.C., Krachter H.U., Lehr C.M., Liebsch M., Mehling A., Mueller-Goymann C., Netzlaff F., Niedorf F., Rubbelke M.K., Schafer U., Schmidt E., Schreiber S., Spielmann H., Vuia A., and Weimer M.The use of reconstructed human epidermis for skin absorption testing: results of the validation study. Altern Lab Anim 36, 161, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Fransson J., Heffler L.C., Linder M.T., and Scheynius A.Culture of human epidermal langerhans cells in a skin equivalent. Br J Dermatol 139, 598, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Regnier M., Patwardhan A., Scheynius A., and Schmidt R.Reconstructed human epidermis composed of keratinocytes, melanocytes and langerhans cells. Med Biol Eng Comput 36, 821, 1998 [DOI] [PubMed] [Google Scholar]
- 27.Auxenfans C., Fradette J., Lequeux C., Germain L., Kinikoglu B., Bechetoille N., Braye F., Auger F.A., and Damour O.Evolution of three dimensional skin equivalent models reconstructed in vitro by tissue engineering. Eur J Dermatol 19, 107, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Gangatirkar P., Paquet-Fifield S., Li A., Rossi R., and Kaur P.Establishment of 3d organotypic cultures using human neonatal epidermal cells. Nat Protoc 2, 178, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Souto L.R., Rehder J., Vassallo J., Cintra M.L., Kraemer M.H., and Puzzi M.B.Model for human skin reconstructed in vitro composed of associated dermis and epidermis. Sao Paulo Med J 124, 71, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mironov V., Visconti R.P., Kasyanov V., Forgacs G., Drake C.J., and Markwald R.R.Organ printing: tissue spheroids as building blocks. Biomaterials 30, 2164, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mironov V., Boland T., Trusk T., Forgacs G., and Markwald R.R.Organ printing: computer-aided jet-based 3d tissue engineering. Trends Biotechnol 21, 157, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Norotte C., Marga F.S., Niklason L.E., and Forgacs G.Scaffold-free vascular tissue engineering using bioprinting. Biomaterials 30, 5910, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roth E.A., Xu T., Das M., Gregory C., Hickman J.J., and Boland T.Inkjet printing for high-throughput cell patterning. Biomaterials 25, 3707, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Xu T., Jin J., Gregory C., Hickman J.J., and Boland T.Inkjet printing of viable mammalian cells. Biomaterials 26, 93, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Bettinger C.J., Weinberg E.J., Kulig K.M., Vacanti J.P., Wang Y.D., Borenstein J.T., and Langer R.Three-dimensional microfluidic tissue-engineering scaffolds using a flexible biodegradable polymer. Adv Mater 18, 165, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borenstein J.T., Weinberg E.J., Orrick B.K., Sundback C., Kaazempur-Mofrad M.R., and Vacanti J.P.Microfabrication of three-dimensional engineered scaffolds. Tissue Eng 13, 1837, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Cooke M.N., Fisher J.P., Dean D., Rimnac C., and Mikos A.G.Use of stereolithography to manufacture critical-sized 3d biodegradable scaffolds for bone ingrowth. J Biomed Mater Res Part B 64B, 65, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Dhariwala B., Hunt E., and Boland T.Rapid prototyping of tissue-engineering constructs, using photopolymerizable hydrogels and stereolithography. Tissue Eng 10, 1316, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Golden A.P., and Tien J.Fabrication of microfluidic hydrogels using molded gelatin as a sacrificial element. Lab Chip 7, 720, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Melchels F.P., Grijpma D., and Feijen J.Photo-crosslinking of functionalised lactide oligomers for the fabrication of osteochondral tissue engineering scaffolds. J Control Release 116, e98, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Lee W., Debasitis J.C., Lee V.K., Lee J.H., Fischer K., Edminster K., Park J.K., and Yoo S.S.Multi-layered culture of human skin fibroblasts and keratinocytes through three-dimensional freeform fabrication. Biomaterials 30, 1587, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Lee W., Pinckney J., Lee V., Lee J.H., Fischer K., Polio S., Park J.K., and Yoo S.S.Three-dimensional bioprinting of rat embryonic neural cells. Neuroreport 20, 798, 2009 [DOI] [PubMed] [Google Scholar]
- 43.Lee W., Lee V., Polio S., Keegan P., Lee J.H., Fischer K., Park J.K., and Yoo S.S.On-demand three-dimensional freeform fabrication of multi-layered hydrogel scaffold with fluidic channels. Biotechnol Bioeng 105, 1178, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Zhao L.L., Lee V.K., Yoo S.S., Dai G.H., and Intes X.The integration of 3-d cell printing and mesoscopic fluorescence molecular tomography of vascular constructs within thick hydrogel scaffolds. Biomaterials 33, 5325, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee Y.-B., Polio S., Lee W., Dai G., Menon L., Carroll R.S., and Yoo S.-S.Bio-printing of collagen and vegf-releasing fibrin gel scaffolds for neural stem cell culture. Exp Neurol 223, 645, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Prunieras M., Regnier M., and Woodley D.Methods for cultivation of keratinocytes with an air-liquid interface. J Invest Dermatol 81, 28s, 1983 [DOI] [PubMed] [Google Scholar]
- 47.Lademann J., Otberg N., Richter H., Meyer L., Audring H., Teichmann A., Thomas S., Knüttel A., and Sterry W.Application of optical non-invasive methods in skin physiology: a comparison of laser scanning microscopy and optical coherent tomography with histological analysis. Skin Res Technol 13, 119, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Pontiggia L., Klar A., Boettcher-Haberzeth S., Biedermann T., Meuli M., and Reichmann E.Optimizing in vitro culture conditions leads to a significantly shorter production time of human dermo-epidermal skin substitutes. Pediatr Surg Int 29, 249, 2013 [DOI] [PubMed] [Google Scholar]
- 49.Yang J., Meyer M., Mueller A.-K., Boehm F., Grose R., Dauwalder T., Verrey F., Kopf M., Partanen J., Bloch W., Ornitz D.M., and Werner S.Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J Cell Biol 188, 935, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yannas I.V., Lee E., Orgill D.P., Skrabut E.M., and Murphy G.F.Synthesis and characterization of a model extracellular matrix that induces partial regeneration of adult mammalian skin. Proc Natl Acad Sci U S A 86, 933, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.