

Significance
Several functions in cells require reductive processes, i.e., the enzymatic catalysis of a transfer of electrons to specific cellular substrates. One major reductive system in the cytosol of human cells depends upon thioredoxin 1 (Trx1), which in turn is kept reduced by thioredoxin reductase 1 (TrxR1) using NADPH. In the present study it is shown that another protein in addition to Trx1, called thioredoxin-related protein of 14 kDa (TRP14), is highly efficient together with TrxR1 in catalyzing reduction of L-cystine or nitric oxide-derivatized cysteine residues. It is also shown that TRP14, in contrast to Trx1, is resistant to inactivation by hydrogen peroxide. These findings reveal that several TrxR1-dependent functions in cells may not be propelled solely by Trx1, but instead relate to activities of TRP14.
Keywords: redox regulation, nitric oxide, sulfur metabolism, oxidative stress
Abstract
Thioredoxin-related protein of 14 kDa (TRP14, also called TXNDC17 for thioredoxin domain containing 17, or TXNL5 for thioredoxin-like 5) is an evolutionarily well-conserved member of the thioredoxin (Trx)-fold protein family that lacks activity with classical Trx1 substrates. However, we discovered here that human TRP14 has a high enzymatic activity in reduction of l-cystine, where the catalytic efficiency (2,217 min−1⋅µM−1) coupled to Trx reductase 1 (TrxR1) using NADPH was fivefold higher compared with Trx1 (418 min−1⋅µM−1). Moreover, the l-cystine reduction with TRP14 was in contrast to that of Trx1 fully maintained in the presence of a protein disulfide substrate of Trx1 such as insulin, suggesting that TRP14 is a more dedicated l-cystine reductase compared with Trx1. We also found that TRP14 is an efficient S-denitrosylase with similar efficiency as Trx1 in catalyzing TrxR1-dependent denitrosylation of S-nitrosylated glutathione or of HEK293 cell-derived S-nitrosoproteins. Consequently, nitrosylated and thereby inactivated caspase 3 or cathepsin B could be reactivated through either Trx1- or TRP14-catalyzed denitrosylation reactions. TRP14 was also, in contrast to Trx1, completely resistant to inactivation by high concentrations of hydrogen peroxide. The oxidoreductase activities of TRP14 thereby complement those of Trx1 and must therefore be considered for the full understanding of enzymatic control of cellular thiols and nitrosothiols.
The redox or nitrosylation state of reactive cysteine (Cys) residues in proteins can affect a multitude of intracellular events, either beneficial or harmful, depending upon biological context (1, 2). Two major cellular systems that control the redox states of Cys residues are the thioredoxin (Trx) and the glutathione (GSH) systems. The Trx system includes isoforms of Trx, Trx reductase (TrxR), and NADPH together with several Trx-dependent enzymes and proteins (3). The GSH/GSH disulfide redox couple is kept reduced by the NADPH-dependent activity of GSH reductase (GR) and donates electrons to isoforms of glutaredoxin (Grx) and other GSH-dependent enzymes (4).
In addition to Trx, many proteins have a Trx fold and a Trx-like active-site sequence. One such protein is thioredoxin-related protein of 14 kDa (TRP14, also known as TXNDC17 or TXNL5), which is an evolutionarily well-conserved cytosolic and widely expressed Trx-fold protein that can be reduced by TrxR1 (5). Its crystal structure, compared with Trx1, shows additional structural features in the active site, thereby explaining its lack of activity with most classical Trx1 protein disulfide substrates including ribonucleotide reductase, insulin, peroxiredoxins, or methionine sulfoxide reductase (5–7). TRP14 was suggested to have evolved to exert specific signaling roles in cells and was identified as a modulator of TNFα/NFκB signaling pathways through interactions with the dynein light chain LC8 protein (6, 8). We previously found that treatment of cells with cisplatin triggered the formation of covalent cross-links between TrxR1 and either Trx1 or TRP14, which suggests that TRP14 is tightly linked to TrxR1 within the cellular context (9). Recently, we also found that TRP14 is able to reactivate oxidized phosphotyrosine phosphatase 1B, thereby indeed implicating specific functions in modulation of cellular signaling pathways (10).
In addition to having general protein disulfide reductase activities, Trx1 is also a denitrosylase for a broad spectrum of nitrosoproteins and nitrosothiols (11, 12). Substrates include S-nitrosocaspase 3 (13, 14), S-nitrosocaspase 8 (15), S-nitrosoglutathione (GSNO) (16, 17), and S-nitrosocysteine (l-CysSNO) (12). Nitrosylation and denitrosylation reactions provide a regulatory mechanism for protein function and are thereby also involved in a variety of cellular signal transduction pathways. For example, S-nitrosylation of caspases can inhibit their activity and thus regulate apoptosis in resting cells (18, 19). A full understanding of NO homeostasis and its pathways is of medical importance because an aberrant formation of nitrosylated proteins has been implicated in a variety of diseases. However, protein denitrosylation is a hitherto less studied part in NO-mediated signaling (20, 21). In addition to Trx1, another enzyme mediating cellular denitrosylation reactions is GSNO reductase (GSNOR). GSNOR is the same enzyme as class III alcohol dehydrogenase, mainly catalyzing denitrosylation of GSNO using NADH as an electron donor (22, 23). In addition, S-denitrosylation activities are supported by protein disulfide isomerase (PDI) (24), carbonyl reductase 1 (25), and lipoic acid (17).
The high intracellular concentrations of GSH are also important in NO metabolism because of facilitated formation of GSNO by reaction of GSH with NO or by denitrosylation of cellular nitrosothiols (20, 26). Because the synthesis of GSH depends upon availability, cellular uptake and reduction of sulfur-containing precursors such as l-cystine, l-cystine homeostasis is also important for GSH functions (27). l-Cystine is taken up into cells using different transport systems, e.g., the oxidative stress-inducible cystine/glutamate antiporter (system 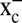 ) (28). The mechanism behind the reduction of l-cystine still has not been fully elucidated, but has been implicated to include GSH itself or also TrxR1-dependent systems (29).
) (28). The mechanism behind the reduction of l-cystine still has not been fully elucidated, but has been implicated to include GSH itself or also TrxR1-dependent systems (29).
In the present study, we wanted to further characterize the enzymatic properties of TRP14, which revealed that the protein is at least as efficient as Trx1 in supporting reduction of specific redox substrates, such as l-cystine. In that assay, TRP14 is a fivefold better substrate for TrxR1 than Trx1 itself and, furthermore, more dedicated as its activity is not diminished in the presence of other Trx1 substrates that are not reduced by TRP14. Furthermore, we discovered that TRP14 is yet another cytosolic oxidoreductase that can catalyze S-denitrosylation reactions.
Results
TRP14 Is an Efficient L-Cystine Reductase.
Like Trx1, TRP14 was earlier shown to be a ubiquitously expressed cytosolic protein (5). Using estimates from immunoblots with recombinant protein standards, we first compared the expression of TRP14 and Trx1 in different cell lines. This showed that Trx1 levels were about two- to threefold higher than those of TRP14, but the two proteins showed different profiles of expression with the highest levels of TRP14 found in A431 cells whereas Trx1 was highest in A549 cells (Fig. 1A).
Fig. 1.
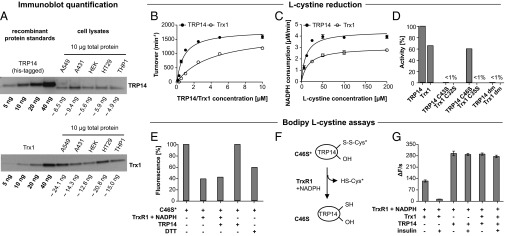
TRP14 and Trx levels differ between cell lines, and TRP14 is an efficient l-cystine reductase. In A, the expression of TRP14 and Trx1 in different cell lines was estimated by immunoblots and standard curves with recombinant proteins as indicated. The calculated amounts of TRP14 or Trx1 per 10 μg of total cellular protein were estimated by densitometry analyses. Kinetic analyses using l-cystine were performed using either varying TRP14/Trx1 (B) or varying l-cystine concentrations (C). In B, the reaction mixtures contained 2 nM TrxR1, 250 μM NADPH and 250 μM l-cystine with turnover calculated with regards to TrxR1, and, in C, 10 nM TrxR1, 250 μM NADPH and 15 μM TRP14/Trx1 were used with NADPH consumption given at the y axis. In D, l-cystine- and TrxR1-coupled NADPH consumption activities under saturating conditions with either TRP14 or Trx1 active-site variants are shown. The reaction mixtures contained 20 μM TRP14/Trx1 variants, 10 nM TrxR1, 250 µM NADPH, and 500 μM l-cystine (n = 3, mean ± SEM). In E–G, fluorescent Bodipy–l-cystine was used and (E) shows fluorescent labeling of TRP14 C46S. Upon incubation with Bodipy–l-cystine, TRP14 C46S (2.5 µM) became fluorescent (C46S*) and thus cysteinylated (the fluorescence was set to 100%). Incubation of this protein with 5 nM TrxR1 and 250 µM NADPH with subsequent desalting resulted in a decrease in fluorescence of the protein, but not with 2.5 µM TRP14 alone. Incubation with 1 mM DTT served as control (n = 2, mean ± SEM). The scheme in F depicts the proposed decysteinylation activity catalyzed by TrxR1 as shown in E. In G, a Bodipy–l-cystine activity assay is shown using 2 nM TrxR1, 250 μM NADPH, 10 μM Bodipy–l-cystine, 160 μM insulin, and 10 μM TRP14/Trx as indicated. The change in fluorescence over time resulting from reduction of Bodipy–l-cystine is plotted on the y axis (n = 3, mean ± SEM).
TRP14 and Trx1 were previously shown to have different substrate specificities, with TRP14 not being able to reduce any classical protein disulfide substrates of Trx1 (5). As TRP14 is expressed at significant levels in cells (Fig. 1A) and may complement the Trx system, we wanted to study the possible reduction of other substrates by TRP14 and compare these to Trx1. We first analyzed hydrogen peroxide (H2O2), hydroxyethyldisulfide (HED), and l-cystine as electron acceptors, which revealed that TRP14 was comparable to Trx1 in reduction of H2O2, about half as efficient as Trx1 with HED, and about twice as active with l-cystine when using 2 µM TRP14 compared with 6 µM Trx1 (Table 1). l-cystine was previously found to be a substrate of Trx1, but not as potent as insulin disulfides (30). Upon closer analysis we found that TRP14 was clearly more potent in l-cystine reduction than Trx1, which was seen varying either protein (Fig. 1B) or substrate (Fig. 1C) concentrations. The kinetic parameters of TrxR1 for either TRP14 or Trx1 in this l-cystine–coupled assay displayed a Km of 0.8 μM for TRP14, compared with 4.3 μM for Trx1, with similar kcat (Table 2). Thus, the catalytic efficiency (kcat/Km) of TrxR1 for TRP14 in this assay was about fivefold higher than that using Trx1, i.e., 2,217 min−1⋅μM−1 for TRP14 compared with 418 min−1⋅μM−1 for Trx1.
Table 1.
Activities of TRP14 and Trx1 with selected substrates
| Turnover (μM NADPH min−1) | ||
| Substrate | TRP14 | Trx1 |
| H2O2, 5 mM | 2.0 ± 0.1 | 2.5 ± 0.4 |
| HED, 2.5 mM | 5.6 ± 0.7 | 9.8 ± 0.5 |
| l-cystine, 250 μM | 14.7 ± 0.7 | 7.4 ± 0.2 |
Substrate concentrations are given in the table, and turnover was determined using either TRP14 (2 µM) or Trx1 (6 µM) with 10 nM TrxR1 and 250 μM NADPH (n = 3, ± SEM). The reactions were carried out in a total volume of 200 μL of TE buffer.
Table 2.
Kinetic parameters of mammalian TrxR1 for TRP14 and Trx1 using L-cystine as ultimate electron acceptor
| Substrate | Km (μM) | kcat (min−1) | kcat/Km (min−1 μM−1) |
| TRP14 | 0.8 ± 0.1 | 1,818 ± 54 | 2,217 ± 233 |
| Trx1 | 4.3 ± 0.6 | 1,749 ± 67 | 418 ± 36 |
The reaction mixture contained 10 nM TrxR1, 250 μM NADPH, and 250 μM l-cystine (n = 3, ± SEM). The reactions were carried out in a total volume of 200 μL of TE buffer.
It was previously shown that the disulfide/dithiol motif formed by Cys43 and Cys46 is the active site of TRP14 (5). Thus, we next analyzed the corresponding Cys-to-Ser mutants of TRP14 using l-cystine as substrate. As expected, the mutant protein with both active-site Cys residues replaced by Ser [double mutant (dm)] was unable to support TrxR1-dependent NADPH oxidation coupled to l-cystine. Surprisingly, however, the C46S monothiol mutant of TRP14 still had substantial activity, whereas the corresponding mutant of Trx1 lacked activity (Fig. 1D). To clarify the reaction mechanism of the C46S variant of TRP14, we developed a fluorescence-based activity assay using Bodipy-labeled l-cystine. The labeled l-cystine is nonfluorescent due to quenching of the two fluorophores, but reduction and thereby separation of the Bodipy fluorophores results in green fluorescence. Importantly, although Bodipy–l-cystine was not as good a substrate for TRP14 as native l-cystine, no background activity was detected with only TrxR1 and NADPH. Bodipy–l-cystine could thereby be used as a probe for analysis of the observed TRP14 C46S activity. Indeed, we found that upon incubation with Bodipy–l-cystine, TRP14 C46S became fluorescent (and was thus cysteinylated). This fluorescence was lowered by incubation with 5 nM TrxR1 and 250 µM NADPH, with or without inclusion of wild-type TRP14 (2.5 µM) or 1 mM DTT, but not using wild-type TRP14 alone (Fig. 1E). These results collectively suggest that TRP14 C46S can be cysteinylated at the remaining Cys43 residue and that reduction of l-cystine by TRP14 and Trx1 occurs through different mechanisms. The disulfide of cysteinylated TRP14 C46S could evidently be a direct substrate for TrxR1, which should explain the activity of TRP14 C46S in l-cystine reduction (see proposed mechanism in Fig. 1F). We next compared the activities of TRP14 and Trx1 in the Bodipy–l-cystine assay and found Trx1 to be about 40% as active as TRP14 (Fig. 1G), in agreement with the results using natural l-cystine as substrate (Fig. 1 B and C). Importantly, upon addition of insulin to this assay as an alternative Trx1 substrate, l-cystine reduction by Trx1 was effectively blocked whereas turnover with TRP14 was not affected (Fig. 1G). This finding suggests that Trx1-mediated l-cystine reduction is inefficient in the presence of other preferred Trx1 substrates whereas, conversely, TRP14 seems to be a more dedicated l-cystine reductase.
Reduction of HED Is Supported by TRP14 and Trx1, but Not if Coupled to GSH.
The disulfide compound HED is commonly used to measure activities of Grx isoforms (31), but shows activity also with either Trx1 or TRP14 in TrxR1-coupled assays (Table 1). Further analyses gave an apparent Km of 3.7 mM for TRP14 with HED compared with 0.5 mM for Trx1 with similar turnover under saturating conditions using 6 µM Trx1 or 2 µM TRP14 (Fig. 2A). It should be noted, however, that, when the HED reduction assay was coupled to GSH, GR, and NADPH, i.e., the classical conditions for Grx activity measurements (31), no activity could be detected with either Trx1 or TRP14 (Fig. 2B). This finding showed that neither Trx1 nor TRP14 can use GSH as electron donor, which could be expected because Grx1 has a GSH-binding pocket lacking in either Trx1 or TRP14 (6, 31, 32).
Fig. 2.

Both TRP14 and Trx1 can reduce HED, but not when coupled to GSH. In A, HED reduction activity coupled to TrxR1 and NADPH is shown. Kinetic analysis with varying HED concentrations was performed according to Michaelis–Menten, and the reaction mixture contained 10 nM TrxR1, 250 μM NADPH, and 6 μM Trx1 or 2 μM TRP14 (n = 3, mean ± SEM). In B, HED reduction coupled to GR, GSH, and NADPH is shown, using standard Grx assay conditions (31). All reaction mixtures contained 1 mM HED, 250 µM NADPH, 1 mM GSH, and 6 µg/mL GR and, in addition, 5 µM of Grx1, Trx1, or TRP14 as indicated (n = 3, mean ± SEM).
Reduction of H2O2 by TRP14 Is, in Contrast to Trx1, Maintained at H2O2 Concentrations Above 5 mM.
A peroxidase activity of TRP14 using H2O2 as substrate up to 10 mM was discovered earlier by others (5). Here, we found that this activity was maintained and increased using very high H2O2 concentrations (up to 40 mM), which could not be seen with Trx1 (Fig. 3A). Inactivation of Trx1 at higher H2O2 concentrations is explained by oxidation of Trx1 into inactive multimers formed through disulfides between structural Cys residues (32, 33). Consequently, Trx1 easily formed DTT-reducible multimers, irrespective of the active-site status, when treated with H2O2 (Fig. 3B, Left). In contrast, both wild-type TRP14 and the active-site double mutant formed a DTT-reducible dimer, but not multimers. Interestingly, the C43S mutant completely lacked aggregation upon oxidation, whereas the C46S mutant displayed strong tendencies to multimerization (Fig. 3B, Right). Again, these reactions with H2O2 revealed different catalytic mechanisms and behavior between Trx1 and TRP14.
Fig. 3.

TRP14 can reduce H2O2 and is in contrast to Trx1 not inactivated by high H2O2 levels. In A, kinetic analyses were performed with varying H2O2 concentrations. The reaction mixture contained 10 nM TrxR1, 250 μM NADPH, and 6 μM Trx1 or 2 μM TRP14 (n = 3, mean ± SEM). In B, 10 µg of prereduced Trx1 (Left) or TRP14 (Right) variants, as indicated, were incubated with 1 mM H2O2 for 20 min and subsequently analyzed on SDS/PAGE with or without reduction of disulfides (±DTT).
TRP14 Has S-Denitrosylase Activity.
Similarly to Trx1 (16), we found that TRP14 has GSNO reductase activity (Fig. 4A). For Trx1, the velocity in GSNO reduction decreased at GSNO concentrations of 50 μM or higher, in agreement with earlier findings (16), whereas no such decrease in activity was seen with TRP14 (Fig. 4A). Determining the release of NO from GSNO using a sensitive NO analyzer demonstrated a time-dependent decrease of GSNO-derived NO upon prior incubation with TrxR1 and NADPH together with either Trx1 or TRP14, but not with the active-site double mutant of TRP14 (Fig. 4B). These results thereby revealed that TRP14 can catalyze S-denitrosylation of GSNO to a similar extent as Trx1. We also found that, coupled to TrxR1 and NADPH, both TRP14 and Trx1 could reduce the complex mixture of nitrosylated proteins in l-CysSNO–treated whole HEK293 cell lysate (molecular mass >30 kDa), whereas incubation with only TrxR1 and NADPH was less efficient (Fig. 4C). Also, the denitrosylation reactions with Trx1 or TRP14 were fully dependent upon TrxR1 and NADPH, illustrating that the activities required enzymatic turnover (Fig. 4C).
Fig. 4.
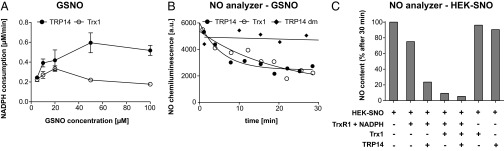
Both Trx1 and TRP14 catalyze denitrosylation of GSNO and S-nitrosoproteins derived from l-CysSNO–treated HEK293-cell lysate (HEK-SNO). In A, GSNO reduction is shown, using GSNO (5–100 μM) incubated with 5 μM of TRP14 or Trx1, together with 250 μM NADPH and 20 nM TrxR1, with NADPH consumption followed at 340 nm over time (n = 3, mean ± SEM). In B, results of TRP14- and Trx1-catalyzed denitrosylation of GSNO measured in a NO analyzer are shown. For this, 10 µM of GSNO were incubated with 5 nM TrxR1, 50 µM NADPH, and, in addition, 5 µM of either Trx1 or wild-type TRP14 at 37 °C, whereupon at the given time points aliquots from the reaction mixture were assessed for NO content in the NO analyzer. A sample containing Cys-to-Ser active-site double mutant TRP14 served as a control. For the analysis of denitrosylation of S-nitrosoproteins derived from l-CysSNO–treated HEK293-cell lysate (C), 2.5 μM of total nitrosylated cellular proteins (as estimated with a GSNO standard curve, molecular mass >30 kDa) were incubated with 5 nM TrxR1 and 100 μM NADPH and, in addition, 5 μM of Trx1 and/or TRP14 at 37 °C. The NO content of each reaction mixture was assessed after ∼30 min and is given relative to the NO content of HEK-SNO (which was set at 100%).
We next studied time-dependent release of NO from S-nitrosylated caspase 3 upon incubation with TrxR1, NADPH, and either TRP14 or Trx1, which displayed comparable activities (Fig. 5A). We also analyzed whether the protease activity of caspase 3 could be modulated by TRP14, as previously shown for Trx1 (13). Indeed, treatment with l-CysSNO strongly inhibited caspase 3, as expected, whereas subsequent incubation with TRP14 reactivated caspase 3 to about 40%, compared with ∼60% for Trx1 (Fig. 5B). We also assessed reactivation of S-nitrosylated cathepsin B, a lysosomal protease known to be inhibited by S-nitrosylation (34, 35), and we found that both Trx1 and TRP14 could reactivate S-nitrosylated cathepsin B, with Trx1 again being more efficient than TRP14 under the conditions of this assay (Fig. 5B).
Fig. 5.

Both Trx1 and TRP14 denitrosylate and thereby reactivate nitrosylated caspase 3 and cathepsin B. (A) TRP14-mediated denitrosylation of Casp3-SNO compared with Trx1 measured with the NO analyzer. Casp3-SNO was incubated with 5 nM TrxR1 and 100 μM NADPH (control) and, in addition, either 5 μM TRP14 or 5 μM Trx1 at 37 °C, and aliquots from the reaction mixture were assessed for NO content in the NO analyzer at the given time points. Effects of l-CysSNO and the Trx/TRP14 system on the activity of caspase 3 and cathepsin B are shown in B. The assays were performed with 20 nM caspase 3/caspase 3-SNO and 150 nM cathepsin B/cathepsin B-SNO with the activity of the nonnitrosylated protein set to 100%. The nitrosylated forms of caspase 3 and cathepsin B were further incubated with 5 nM TrxR1 and 100 µM NADPH and, in addition, either 5 µM Trx1 or TRP14. Controls lacking TrxR1 and NADPH were included only for caspase 3-SNO (n = 2, mean ± SEM).
Asking whether knockdown or overexpression of TRP14 or Trx1 would affect the total NADPH-dependent, TrxR1-linked l-cystine reduction or S-denitrosylation activities in cellular extracts, we found that this was indeed the case (Fig. 6).
Fig. 6.

Knockdown and overexpression of either TRP14 or Trx1 affect total l-cystine reduction capacity and S-nitrosoprotein levels in crude HEK293 cell lysates. In A, the relative protein levels of Trx1, TRP14, and TrxR1 upon shRNA-mediated knockdown or overexpression are shown with respective transfection controls as indicated. Protein levels as shown were determined with immunoblotting, using GAPDH in the same samples as loading controls. The blots are representative for two independent experiments. The corresponding cell lysates were subsequently used for a l-cystine reduction activity assay, as shown in B. Only TrxR1-dependent l-cystine reduction is shown, with background controls lacking TrxR1 and NADPH subtracted (n = 2, mean ± SEM). In C, the corresponding cells were treated in culture with l-CysSNO, and thereafter lysates were prepared and assessed for nitrosoprotein content using the DAN assay by spectrophotometrically monitoring DAN conversion to fluorescent 2,3-naphthyltriazole. The fluorescence of nitrosoproteins from each corresponding transfection control was set to 100% (n = 2, mean ± SEM).
Discussion
Previous work by others showed that TRP14 is a ubiquitously expressed Trx-fold protein that can be reduced by TrxR1 (5), but efficient substrates for TRP14 have not yet been identified. Our present study suggests that TRP14 may have redox active roles with importance for l-cystine metabolism and NO signaling. We show here, for the first time to our knowledge, that TRP14 can be at least as active as Trx1 as a substrate for TrxR1, provided that it is coupled to a suitable electron acceptor. Also, TRP14 was found to be a more dedicated l-cystine reductase than Trx1 because its activity was not affected by the presence of another efficient Trx1 substrate such as insulin. We thus suggest that TRP14 should henceforth be considered as an efficient cytosolic TrxR1-dependent oxidoreductase acting in parallel with Trx1.
The fact that TRP14 efficiently reduces l-cystine should be considered for the intracellular l-cystine metabolism and thus also for GSH synthesis. After cellular uptake via the L-cystine/glutamate antiporter (system 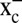 ), l-cystine needs to be reduced for use in GSH synthesis (36). It has been suggested that GSH itself is the main reducing entity in the cytosol, but it was shown that, under conditions of low GSH, TrxR1 clearly contributes to intracellular l-cystine reduction (29, 37). Furthermore, the reduction of l-cystine by GSH is reported to be too slow to be adequate for the required amounts of cysteine in the cell (38). The same study determined the intracellular l-cystine concentration to be 30 μM in HT29 cells (38), a concentration where TRP14 showed twice the activity compared with Trx1 in the absence of other Trx1 substrates (Fig. 1C). Considering that 18% of the total cytosolic l-cystine reduction in rat liver was suggested to have been due to Trx1 (39), we hypothesize that instead TRP14 may contribute to a major part of this cytosolic l-cystine reduction, which should be an evident focus for future studies.
), l-cystine needs to be reduced for use in GSH synthesis (36). It has been suggested that GSH itself is the main reducing entity in the cytosol, but it was shown that, under conditions of low GSH, TrxR1 clearly contributes to intracellular l-cystine reduction (29, 37). Furthermore, the reduction of l-cystine by GSH is reported to be too slow to be adequate for the required amounts of cysteine in the cell (38). The same study determined the intracellular l-cystine concentration to be 30 μM in HT29 cells (38), a concentration where TRP14 showed twice the activity compared with Trx1 in the absence of other Trx1 substrates (Fig. 1C). Considering that 18% of the total cytosolic l-cystine reduction in rat liver was suggested to have been due to Trx1 (39), we hypothesize that instead TRP14 may contribute to a major part of this cytosolic l-cystine reduction, which should be an evident focus for future studies.
Considering that Trx1 is susceptible to inhibitory oxidation by high concentrations of oxidants, as found here and also shown earlier (33, 40), it is notable that TRP14 was resistant to such inhibition. The intracellular removal of H2O2 in cells is mediated mainly by the cytosolic peroxiredoxins using reducing equivalents provided by Trx, so a direct peroxidase activity of Trx is not likely (41). Because TRP14 does not reduce peroxiredoxins, the results rather suggest a regulating mechanism. The concentrations of H2O2 within the millimolar range used here are unlikely to represent physiologic values (42), but indicate that TRP14 could function under conditions where Trx1 is inactivated by oxidation.
The S-denitrosylation activities of TRP14 are noteworthy, as they may mediate TrxR1-dependent modulation of NO metabolism and NO-related signaling. As mentioned, denitrosylation reactions can be catalyzed by Trx1, GSNOR (23), and PDI (24), with GSNOR being the major enzyme for NADH-dependent metabolism of GSNO (22, 43). The denitrosylation of GSNO by Trx1 has been reported earlier (17), and here we hypothesize from our data that cellular TrxR1-dependent, and thereby NADPH-dependent, denitrosylation mechanisms, can be mediated by either Trx1 or TRP14.
Beyond the low-molecular-weight nitrosothiol GSNO, more than 1,000 S-nitrosylated cellular proteins have been implicated in cellular processes or diseases (44). Our findings with HEK293 cell-derived S-nitrosoproteins revealed that both Trx1 and TRP14 can denitrosylate a large part of such nitrosothiol (SNO) functions. Similar results were earlier obtained with Trx1 using S-nitrosoproteins from either HepG2 cells (12) or Jurkat cells (11) as substrates. Specifically, Trx1 was shown earlier to reduce S-nitrosylated caspase 3 with regeneration of protease activity (13, 14), which was also found in the present study and replicated also with TRP14, when coupled to TrxR1 and NADPH. An earlier study could not attribute any caspase 3-SNO denitrosylation activity to TRP14 (13), but because TrxR1 was shown to play significant roles in regenerating nitrosylated caspase 3 in cells (13), our data suggest that TrxR1-dependent denitrosylation reactions may indeed occur through the actions of either Trx1 or TRP14. Because we found that different cells have different proportions of TRP14 vs. Trx1 expression and because Trx1 activities may be diverted to many different Trx1 substrates, we propose that the extent of denitrosylase activity that is provided by TRP14 will depend upon the overall cellular content and activities of the complete Trx system. This notion is also supported by results of the knockdown and overexpression experiments performed here. To understand the overall cellular impact of TRP14 vs. Trx1 activities in different reductive pathways of diverse living cells will, however, require further studies.
To conclude, we here provide evidence that TRP14 is a TrxR1-dependent reductase that can efficiently reduce both l-cystine and S-nitrosothiols. Thus, TRP14 should be considered as a unique cytosolic member of the Trx system with specialized redox activities complementing those of Trx1.
Materials and Methods
Details about cloning, protein purification, immunoblot analyses, cell culture, and cell transfection are provided in SI Material and Methods.
Cloning, Mutagenesis, and Expression of Human TRP14 and Trx1.
The ORF of human TRP14 (cDNA clone from imaGenes) was amplified and inserted into a pET20b vector using standard molecular cloning techniques, thereby introducing an N-terminal His-tag. This and a plasmid for wild-type human Trx1 (33) was used to insert the active-site mutations using PCR-mediated site-directed mutagenesis. All final plasmids were transformed into Escherichia coli BL21 (DE3) competent cells (Invitrogen), which were used to express the respective proteins. Purification of all his-tagged proteins was performed using nickel affinity chromatography. Details about the purification procedure are given in SI Material and Methods.
Enzyme Kinetic Assays.
Previous protocols for Trx (45) and Grx (31) kinetic assays were applied to 96-well microtiter plates, and enzymatic activities were determined by measuring NADPH consumption in 200 μL TE buffer (50 mM Tris⋅HCl, 2 mM EDTA, pH 7.5) with protein concentrations as indicated in each experiment using a VersaMax microplate reader (Molecular Devices). For fluorescent activity assays, Bodipy-labeled l-cystine (Invitrogen) was used as a substrate, and fluorescence changes over time were determined using a Victor3 plate reader (Perkin-Elmer) with 405 nm of excitation and 520-nm emission filters. Specific details about all kinetic measurements are given in SI Material and Methods.
Incubation of Trx1 or TRP14 with H2O2 and SDS/PAGE Analyses.
Trx1 and TRP14 preparations were reduced using incubation for 30 min at ∼20 °C with 20-fold molar excess of DTT, whereupon DTT was removed using Spin Desalting columns (Thermo Scientific). The reduced proteins (10 µg) were then incubated with 1 mM H2O2 for 20 min at ∼20 °C and thereafter directly analyzed on SDS/PAGE (Invitrogen, ±25 mM DTT, as indicated).
S-Nitrosylation of Proteins.
For denitrosylation experiments, HEK-cell–derived proteins were first filtered using ultrafiltration columns [30 kDa molecular weight cutoff (MWCO), Millipore] to remove low-molecular-weight cell components and were subsequently incubated with 1 mM l-CysSNO for 20 min at ∼20 °C, followed by removal of the nitrosylation agent using ultrafiltration columns (30 kDa MWCO, Millipore). Pure caspase 3 or cathepsin B were nitrosylated with 50 µM l-CysSNO for 20 min at ∼20 °C in 0.1 M phosphate buffer containing 0.2 mM EDTA followed by removal of the nitrosylation agent using ultrafiltration columns (Millipore).
Assessment of Denitrosylation Activities.
Nitrosoproteins were assessed using either a nitric oxide analyzer (12) or the 2,3-diaminonaphthalene (DAN) assay (46). To assess denitrosylation of caspase 3 and cathepsin B, fluorescence activity assays using specific substrates coupled to 7-amino-4-methylcoumarin were used (47). Details about all assays are given in SI Material and Methods.
Supplementary Material
Acknowledgments
The authors thank Dr. Olle Rengby and Dr. Tomas Gustafsson for assistance in cloning and protein expression and Carina Nihlén for excellent technical assistance with the NO analyzer (all from the Karolinska Institutet). This study was funded by grants from the Swedish Cancer Society (to E.S.J.A. and A.H.), the Swedish Research Council (Medicine), and the Karolinska Institutet.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317320111/-/DCSupplemental.
References
- 1.Nakamura H, Nakamura K, Yodoi J. Redox regulation of cellular activation. Annu Rev Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- 2.Foster MW, McMahon TJ, Stamler JS. S-nitrosylation in health and disease. Trends Mol Med. 2003;9(4):160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 3.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267(20):6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 4.Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780(11):1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Jeong W, Yoon HW, Lee SR, Rhee SG. Identification and characterization of TRP14, a thioredoxin-related protein of 14 kDa. New insights into the specificity of thioredoxin function. J Biol Chem. 2004;279(5):3142–3150. doi: 10.1074/jbc.M307932200. [DOI] [PubMed] [Google Scholar]
- 6.Woo JR, et al. Structural basis of cellular redox regulation by human TRP14. J Biol Chem. 2004;279(46):48120–48125. doi: 10.1074/jbc.M407079200. [DOI] [PubMed] [Google Scholar]
- 7.Jeong W, Jung Y, Kim H, Park SJ, Rhee SG. Thioredoxin-related protein 14, a new member of the thioredoxin family with disulfide reductase activity: Implication in the redox regulation of TNF-alpha signaling. Free Radic Biol Med. 2009;47(9):1294–1303. doi: 10.1016/j.freeradbiomed.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 8.Jeong W, Chang TS, Boja ES, Fales HM, Rhee SG. Roles of TRP14, a thioredoxin-related protein in tumor necrosis factor-alpha signaling pathways. J Biol Chem. 2004;279(5):3151–3159. doi: 10.1074/jbc.M307959200. [DOI] [PubMed] [Google Scholar]
- 9.Prast-Nielsen S, Cebula M, Pader I, Arnér ES. Noble metal targeting of thioredoxin reductase–covalent complexes with thioredoxin and thioredoxin-related protein of 14 kDa triggered by cisplatin. Free Radic Biol Med. 2010;49(11):1765–1778. doi: 10.1016/j.freeradbiomed.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Dagnell M, et al. Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGF-β receptor tyrosine kinase signaling. Proc Natl Acad Sci USA. 2013;110(33):13398–13403. doi: 10.1073/pnas.1302891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benhar M, Thompson JW, Moseley MA, Stamler JS. Identification of S-nitrosylated targets of thioredoxin using a quantitative proteomic approach. Biochemistry. 2010;49(32):6963–6969. doi: 10.1021/bi100619k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sengupta R, et al. Thioredoxin catalyzes the denitrosation of low-molecular mass and protein S-nitrosothiols. Biochemistry. 2007;46(28):8472–8483. doi: 10.1021/bi700449x. [DOI] [PubMed] [Google Scholar]
- 13.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320(5879):1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sengupta R, Billiar TR, Atkins JL, Kagan VE, Stoyanovsky DA. Nitric oxide and dihydrolipoic acid modulate the activity of caspase 3 in HepG2 cells. FEBS Lett. 2009;583(21):3525–3530. doi: 10.1016/j.febslet.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sengupta R, Billiar TR, Kagan VE, Stoyanovsky DA. Nitric oxide and thioredoxin type 1 modulate the activity of caspase 8 in HepG2 cells. Biochem Biophys Res Commun. 2010;391(1):1127–1130. doi: 10.1016/j.bbrc.2009.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nikitovic D, Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem. 1996;271(32):19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 17.Stoyanovsky DA, et al. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J Am Chem Soc. 2005;127(45):15815–15823. doi: 10.1021/ja0529135. [DOI] [PubMed] [Google Scholar]
- 18.Mannick JB, et al. S-nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154(6):1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rössig L, et al. Nitric oxide inhibits caspase-3 by S-nitrosation in vivo. J Biol Chem. 1999;274(11):6823–6826. doi: 10.1074/jbc.274.11.6823. [DOI] [PubMed] [Google Scholar]
- 20.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10(10):721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 21.Anand P, Stamler JS. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J Mol Med (Berl) 2012;90(3):233–244. doi: 10.1007/s00109-012-0878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu L, et al. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410(6827):490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 23.Jensen DE, Belka GK, Du Bois GC. S-nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem J. 1998;331(Pt 2):659–668. doi: 10.1042/bj3310659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sliskovic I, Raturi A, Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem. 2005;280(10):8733–8741. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- 25.Bateman RL, Rauh D, Tavshanjian B, Shokat KM. Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J Biol Chem. 2008;283(51):35756–35762. doi: 10.1074/jbc.M807125200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clancy R, Cederbaum AI, Stoyanovsky DA. Preparation and properties of S-nitroso-L-cysteine ethyl ester, an intracellular nitrosating agent. J Med Chem. 2001;44(12):2035–2038. doi: 10.1021/jm000463f. [DOI] [PubMed] [Google Scholar]
- 27.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134(3):489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 28.Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (−): Cystine supplier and beyond. Amino Acids. 2012;42(1):231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 29.Mandal PK, et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. 2010;285(29):22244–22253. doi: 10.1074/jbc.M110.121327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmgren A. Reduction of disulfides by thioredoxin. Exceptional reactivity of insulin and suggested functions of thioredoxin in mechanism of hormone action. J Biol Chem. 1979;254(18):9113–9119. [PubMed] [Google Scholar]
- 31.Holmgren A, Åslund F. Glutaredoxin. Methods Enzymol. 1995;252:283–292. doi: 10.1016/0076-6879(95)52031-7. [DOI] [PubMed] [Google Scholar]
- 32.Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 33.Ren X, Björnstedt M, Shen B, Ericson ML, Holmgren A. Mutagenesis of structural half-cystine residues in human thioredoxin and effects on the regulation of activity by selenodiglutathione. Biochemistry. 1993;32(37):9701–9708. doi: 10.1021/bi00088a023. [DOI] [PubMed] [Google Scholar]
- 34.Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets. 2013;17(3):281–291. doi: 10.1517/14728222.2013.740461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamler JS, et al. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA. 1992;89(1):444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banjac A, et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27(11):1618–1628. doi: 10.1038/sj.onc.1210796. [DOI] [PubMed] [Google Scholar]
- 37.Banerjee R. Redox outside the box: Linking extracellular redox remodeling with intracellular redox metabolism. J Biol Chem. 2012;287(7):4397–4402. doi: 10.1074/jbc.R111.287995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones DP, et al. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J. 2004;18(11):1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- 39.Mannervik B, Axelsson K, Sundewall AC, Holmgren A. Relative contributions of thioltransferase-and thioredoxin-dependent systems in reduction of low-molecular-mass and protein disulphides. Biochem J. 1983;213(2):519–523. doi: 10.1042/bj2130519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashemy SI, Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J Biol Chem. 2008;283(32):21890–21898. doi: 10.1074/jbc.M801047200. [DOI] [PubMed] [Google Scholar]
- 41.Rhee SG, et al. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17(2):183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 42.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45(5):549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Liu L, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116(4):617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 44.Seth D, Stamler JS. The SNO-proteome: Causation and classifications. Curr Opin Chem Biol. 2011;15(1):129–136. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnér ESJ, Holmgren A. 2000. Measurement of thioredoxin and thioredoxin reductase. Current Protocols in Toxicology, eds Maines M, Costa L, Reed D, Sassa S (John Wiley & Sons, Inc., New York), pp 7.4.1–7.4.14.
- 46.Wink DA, Kim S, Miles A, Jourd’heuil D, Grisham MB. 2001. Methods for distinguishing nitrosative and oxidative chemistry of reactive nitrogen oxide species derived from nitric oxide. Current Protocols in Toxicology, ed Mahin D. Maines (John Wiley & Sons, Inc., New York), Chapter 10:Unit 10 18.
- 47.Pratt MR, Sekedat MD, Chiang KP, Muir TW. Direct measurement of cathepsin B activity in the cytosol of apoptotic cells by an activity-based probe. Chem Biol. 2009;16(9):1001–1012. doi: 10.1016/j.chembiol.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.