

Significance
The recently emerged Middle East Respiratory Syndrome coronavirus (MERS-CoV) causes severe respiratory disease with ∼43% mortality. There is no licensed vaccine or antiviral for MERS. Here we identified seven human neutralizing Abs (nAbs) against MERS-CoV. These nAbs bind to three epitope groups in the viral Spike protein–receptor interface, blocking virus attachment. Five residues in the viral receptor-binding domain critical for neutralization escape were identified. Further study indicated that four of five mutations not only confer neutralization resistance but also impair receptor binding and viral fitness. This panel of nAbs offers the possibility of developing human mAb-based immunotherapy.
Keywords: IGHV1–69, biodefense, emerging pathogen, zoonosis, humoral immunity
Abstract
The newly emerging Middle East Respiratory Syndrome coronavirus (MERS-CoV) causes a Severe Acute Respiratory Syndrome-like disease with ∼43% mortality. Given the recent detection of virus in dromedary camels, zoonotic transfer of MERS-CoV to humans is suspected. In addition, little is known about the role of human neutralizing Ab (nAb) pressure as a driving force in MERS-CoV adaptive evolution. Here, we used a well-characterized nonimmune human Ab-phage library and a panning strategy with proteoliposomes and cells to identify seven human nAbs against the receptor-binding domain (RBD) of the MERS-CoV Spike protein. These nAbs bind to three different epitopes in the RBD and human dipeptidyl peptidase 4 (hDPP4) interface with subnanomolar/nanomolar binding affinities and block the binding of MERS-CoV Spike protein with its hDPP4 receptor. Escape mutant assays identified five amino acid residues that are critical for neutralization escape. Despite the close proximity of the three epitopes on the RBD interface, escape from one epitope did not have a major impact on neutralization with Abs directed to a different epitope. Importantly, the majority of escape mutations had negative impacts on hDPP4 receptor binding and viral fitness. To our knowledge, these results provide the first report on human nAbs against MERS-CoV that may contribute to MERS-CoV clearance and evolution. Moreover, in the absence of a licensed vaccine or antiviral for MERS, this panel of nAbs offers the possibility of developing human mAb-based immunotherapy, especially for health-care workers.
Middle East Respiratory Syndrome coronavirus (MERS-CoV), a newly emergent subgroup C betacoronavirus, was first isolated in the Arabian Peninsula in 2012 (1). Similar to the Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) that emerged in China in 2002, MERS-CoV causes severe respiratory tract infection, often in the lower respiratory tract and occasionally accompanied by renal disease (1). As of February 28, 2014, 184 cases with 80 deaths have been confirmed in 10 countries in the Middle East, Europe, and North Africa (www.who.int/csr/don/2014_02_28/en/). Although the human-to-human transmission rate is mild to moderate at the moment (2, 3), the increasing number of person-to-person transmissions raises concern for a more widespread regional outbreak or even global spread by international travelers, as occurred with SARS-CoV in 2002–2003. Limited information exists on the mechanisms that confer increased human-to-human transmission of MERS-CoV (4). However, mutational adaptation of the SARS-CoV Spike (S) protein for its receptor, angiotensin-converting enzyme 2 (ACE2) was a positive selection factor after zoonotic transfer to humans (5, 6).
Phylogenetic analysis indicates that MERS-CoV is closely related to CoVs detected in Tylonycteris pachypus and Pipistrellus abramus bats in China, Nyctinomops laticaudatus bats in Mexico, and Nycteris cf. gambiensis bats in Ghana and Europe (7–9). An ∼190-bp nucleotide fragment that was genetically identical to the RNA-dependent RNA polymerase of MERS-CoV was detected in Taphozous perforatus bat specimens in the vicinity of the index case in Saudi Arabia (10). Two independent serological surveys of livestock found that dromedary camels had a high prevalence of neutralizing Abs (nAbs) against MERS-CoV (11, 12). Recently, MERS-CoV has been identified from dromedary camels on a farm associated with two human cases, but the transmission patterns remain unclear (13). More recently, a study detected Abs in all 151 samples of dromedary camel serum obtained from the United Arab Emirates in 2003, indicating that MERS-CoV or closely related CoVs existed in the United Arab Emirates long before the first human MERS cases (14, 15). A screen of cell lines derived from livestock and peridomestic small mammals on the Arabian Peninsula revealed that only ungulates such as goats and camels showed efficient replication of MERS-CoV (16). These findings suggest that bats and camels may play an important role in MERS-CoV transmission and that the range of species that can be infected with MERS-CoV may be even broader than currently known (17).
The coronavirus S protein is a class I membrane fusion protein that represents the major envelope protein on the surface of CoVs. The S protein presents as a trimer and mediates receptor binding, membrane fusion, and virus entry. S also is the major target for nAbs (18). It has been reported that patients infected with MERS-CoV generated S protein-specific nAbs (19, 20). The cellular receptor for MERS-CoV has been identified as dipeptidyl peptidase 4 (DPP4, CD26), which is conserved across many species (21). The receptor-binding domain (RBD) of the virus S protein in complex with human DPP4 (hDPP4) has been characterized (22, 23).
Although MERS-CoV has a lower reproduction number (R0) than SARS-CoV (0.69 vs. 0.80) (2), it has a much higher mortality rate (43% vs. 10%). Currently, no licensed vaccines or antivirals are available for the prevention or treatment of MERS. Combination treatment with IFN-α2b and ribavirin can moderate the host response and has been reported to improve clinical outcomes in MERS-CoV–infected rhesus macaques (24). MERS-CoV S protein vaccines based on modified vaccinia virus Ankara or Venezuelan Equine Encephalitis replicon particles and purified RBD can induce virus nAbs in mouse models (25–27). However, results of human studies have not been reported. Thus, an urgent medical need remains for the targeted prophylaxis and treatment of MERS.
Human Ab engineering is a powerful tool that has been used for both discovery and therapeutic applications. We and others have proposed previously that human mAbs could be used in a outbreak setting for the prophylaxis and early treatment of emerging viral pathogens (28, 29). However, obtaining timely access to biological specimens from infected patients as a source of B cells for targeted selection or Ab-phage library construction is often challenging and can delay the discovery process (30–33). These restrictions have led us to use an ultra-large nonimmune human Ab-phage display library as a resource for the isolation of human nAbs to several emerging pathogens (29, 34, 35). With this Ab library resource and a unique panning strategy, we report the isolation and characterization of seven human nAbs that bind to three different epitopes at the MERS-CoV RBD–hDPP4 interface. We also investigated nAb-driven virus evolution and identified residues on the RBD that are critical for neutralization escape. These studies provide insight into the human nAb response that appears to impact MERS-CoV fitness and evolution. In addition, this panel of nAbs offers the possibility of developing a human mAb-based immunotherapy for the prevention and treatment of MERS.
Results
Identification of Anti-MERS Spike Phage Abs by Sequential Spike-Containing Paramagnetic Proteoliposome and Spike-Expressing 293T Cell Panning.
Purified Spike-containing paramagnetic proteoliposomes (S-PMPLs) and Spike-expressing 293 T cells (S-293T) were used to select Abs from the Mehta I/II nonimmune human single-chain variable domain fragment (scFv)-phage libraries. After two rounds of selection with S-PMPLs and one subsequent round of selection with S-293T cells, a total of 1,344 clones were screened to verify their binding to S1 and S2 fused with constant region fragment of human IgG (hFc) (Fig. S1) by ELISA or meso-scale discovery (MSD). Seventy-nine clones were positive against S1-hFc, accounting for all the positive clones binding to S-293T cells, as confirmed by flow cytometric analysis. Further sequencing indicated that the 79 positive clones represented seven unique anti-S1 scFvs (1E9, 1F8, 3A1, 3B12, 3C12, 3B11, and M14D3). Fig. 1 shows the amino acid sequences of these clones. Three different germ-line sequences from the variable region of the heavy chain (VH) and six different germ-line sequences from the variable region of the light chain (VL) are represented. Five of seven VH chains belong to one gene family, IGHV1-69 (four 06 alleles and one 09 allele). Remarkably, these VH segments show very low levels of somatic hypermutation (SHM) (mean 4.3 ± 4.2), ranging from zero (3B12, 3B11) to only five (3A1, 3C12) amino acid substitutions. 1E9 and 1F8 Abs use VH3-30 and VH1-3 and have 10 and 9 substitutions, respectively. The lengths of the VH complementarity-determining region 3 (CDR3) for these seven Abs vary from 11 to 19 amino acids. The VL genes are more diverse. Four VLs use κ chains, and three use λ chains. However, they also show a low level of SHM (mean 5.3 ± 2.4).
Fig. 1.
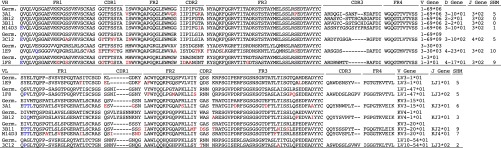
Amino acid sequences of variable regions of anti–MERS-CoV Spike mAbs. Framework regions 1–4 (FR1–4) and CDR1–3 for VH and VL are shown. FR, CDR, and germ line are defined according to the International Immunogene Tics database. Hyphens denote gaps. SHMs are highlighted in red. Because PCR priming may bias the first six amino acids of rearranged VH and VL segments, these amino acids were not included in SHM analysis and are colored in blue. Germ, germ-line gene.
S1 Domain Epitope Mapping and Binding Competition.
To delineate the S1 epitopes of these Abs more precisely, the S1 domain was expressed as three fragments: S1(21–358), S1(349–751), and S1(349–590) (Fig. S1). Each fragment was designed with an N-terminal Flag tag and a C-terminal hFc tag. Binding detection with Octet showed that all seven scFvFcs recognized S1(349–751)-hFc and S1(349–590)-hFc but not S1(21–358)-hFc (Fig. S2). These results demonstrate that the epitopes of all seven Abs lie within the 349–590 amino acid fragment of the S protein, which contains the RBD of MERS-CoV (36).
To determine whether these Abs recognize different epitopes, binding competition assays were performed. An anti-Flag biosensor capturing S1(349–590)-hFc initially was saturated with one Ab, and additional binding with another Ab was evaluated. The results indicate that 1E9 can block the binding of 1F8 and 3A1 completely and can block 3B12 and M14D3 partially but does not affect the additional binding of 3B11 and 3C12 (Fig. S3A). Abs 1F8, 3A1, and 3B12 can block the binding of all other Abs to S1(349–590) (Fig. S3 B–D). Additionally, 3B11, 3C12, and M14D3 can block the binding of 1F8, 3A1, and 3B12, but not 1E9, to S1(349–590) (Fig. S3 E–G). The results indicate that the seven Abs recognize at least three distinct epitope groups, although Ab-RBD cocrystallization will be necessary to determine the precise atomic details of the epitopes and the Ab binding orientations. Ab 1E9 recognizes one unique epitope (group 1); 1F8, 3A1, and 3B12 recognize another epitope or overlapping epitopes close to each other (group 2); and 3B11, 3C12, and M14D3 recognize a third distinct epitope or group of epitopes (group 3). In addition, the ability of epitope 2 Abs to block RBD binding of all Abs suggests that epitope 2 most likely is located centrally, whereas epitopes 1 and 3 are flanking, a conclusion that also is supported by neutralization escape data with MERS-CoV (described below).
Ab Binding Affinity and Neutralization Activity of Anti-Spike scFvFcs and IgGs.
Binding rate constants [equilibrium dissociation constant (Kd), Kon, and Koff] of each scFvFc and their fully converted IgG1 forms to S1(349–590) were measured by bio-layer interferometry (BLI) using an Octet RED96 (Fig. S4). All Abs had nanomolar to subnanomolar binding affinity to S1(349–590) (Table 1). Although it is uncharacteristic in our hands to see a significant loss of binding affinity on scFvFc-to-IgG1 format changes, conversion of three of the seven scFvFcs (3A1, 3B12, and M14D3) to IgG1 resulted in ∼11- to 34-fold higher Kd values, which mostly were attributable to faster Koff rates. For the other four Abs, the IgG1 forms had lower Kd values than their scFvFc counterparts and therefore had higher binding affinities.
Table 1.
Kinetic rates and binding affinity of anti–MERS-CoV Abs
| Ab | scFvFc | IgG | IgG/scFvFc* | ||||||
| Kd, M | Kon, 1/Ms | Koff, 1/s | Kd, M | Kon, 1/Ms | Koff, 1/s | Kd | Kon | Koff | |
| 1E9 | 4.13E-10 | 7.91E+05 | 3.26E-04 | 2.61E-10 | 8.75E+05 | 2.28E-04 | 0.63 | 1.11 | 0.70 |
| 1F8 | 1.58E-09 | 6.68E+05 | 1.05E-03 | 1.11E-09 | 6.52E+05 | 7.22E-04 | 0.70 | 0.98 | 0.69 |
| 3B12 | 1.76E-10 | 1.16E+06 | 2.04E-04 | 2.24E-09 | 2.09E+06 | 4.68E-03 | 12.73 | 1.80 | 22.94 |
| 3A1 | 2.25E-10 | 5.04E+05 | 1.13E-04 | 2.43E-09 | 1.68E+06 | 4.08E-03 | 10.80 | 3.33 | 36.11 |
| 3C12 | 2.01E-09 | 4.85E+05 | 9.76E-04 | 4.44E-10 | 2.82E+06 | 1.25E-03 | 0.22 | 5.81 | 1.28 |
| 3B11 | 1.35E-10 | 1.21E+06 | 1.64E-04 | 5.70E-11 | 1.24E+06 | 7.06E-05 | 0.42 | 1.02 | 0.43 |
| M14D3 | 4.74E-11 | 1.58E+05 | 7.51E-06 | 1.59E-09 | 1.61E+05 | 2.56E-04 | 33.54 | 1.02 | 34.09 |
The kinetic constants were obtained by global analysis using a 1:1 binding model using Fortebio Data Analysis 7.0 software.
Ratio of each rate.
We then tested the neutralization activities of these Abs on hDPP4-expressing 293T cells (293T-hDPP4) with MERS-CoV S pseudotyped lentiviruses (VLPs) carrying a luciferase reporter gene. Although all anti–MERS-S1 Abs can neutralize MERS pseudovirus infection specifically with varying efficiency, 3B11 exhibited the best pseudovirus neutralization activity in both the scFvFc and IgG1 formats (Fig. 2 A and B). To investigate the possible effect of S density on the neutralization potential of the Abs, we compared pseudovirus neutralization with the neutralization of live MERS-CoV infection of Vero cells using plaque reduction neutralization tests (PRNT50). The results indicated that all scFvFcs strongly neutralized MERS-CoV with varying degrees of efficiency. Abs 3B11, 3A1, 3B12, and 3C12 were very strong neutralizers, with 50% inhibitory concentration (IC50) values ranging from 1.25 to 2 µg/mL. The other three scFvFcs, 1E9, M14D3, and 1F8, neutralized the virus with IC50 values of 3.21, 4.3, and 6.27 µg/mL, respectively (Table S1). At concentrations ≥20 µg/mL, six of seven scFvFcs neutralized MERS-CoV completely (Fig. 2C). Three IgG1 Abs also were tested for live virus neutralization; 3B11 had a lower IC50 value than 1F8 and 3A1 IgGs (Table S1). The anti-SARS S mAb 80R did not neutralize MERS-CoV (Fig. 2D) (35).
Fig. 2.
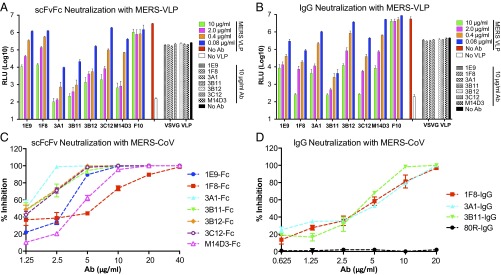
Neutralization with MERS-VLPs (A and B) or live virus (C and D). Serially diluted scFvFc (A) and IgG1 (B) were tested with MERS-VLPs. For the pseudovirus neutralization assay, anti-influenza HA mAb (F10) was used as a negative Ab control, and VSV-G pseudotyped virus was used as pseudovirus control for neutralization specificity. Seven scFvFcs (C) and three IgG1s (D) were tested by PRNT50 assay with live MERS-CoV. The anti-SARS S mAb 80R was used as a negative Ab control.
For pseudovirus neutralization, 1F8, 1E9, 3B11, and 3C12 IgG1 had neutralization activities similar to those of their scFvFcs, in agreement with their similar binding affinities, whereas 3A1, 3B12, and M14D3 IgGs had lower neutralization activities than their scFvFcs, in agreement with their lower binding affinities (Fig. 2B and Table 1). Thus, as expected, virus neutralization potency was positively correlated with nAb binding affinity, with 3B11 being the most potent nAb in both formats.
Mechanism(s) of MERS-CoV Neutralization.
We next explored the mechanism(s) of neutralization by investigating whether these Abs could block hDPP4 binding to the RBD and vice versa. Binding competition between hDPP4 and scFvFcs was conducted with Octet with a technique similar to that used to detect the competition between each Ab (Fig. S3). As shown in Fig. 3A, all scFvFcs could block hDPP4 binding to S1(349–590), whereas preloading of control scFvFc (F10) did not affect hDPP4 binding to S1(349–590). Similarly, hDPP4 also could block all seven scFvFcs from binding to S1(349–590) (Fig. 3B).
Fig. 3.
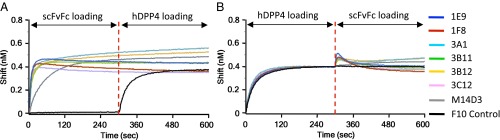
Competition for binding to the MERS S RBD between anti-S1 Abs and hDPP4. Immobilized S1(349–590)-hFc (RBD-hFc) was saturated with 100 nM of scFvFc (A) or hDPP4 (B), and the RBD sensors then were exposed to hDPP4 (A) or scFvFc (B). The binding properties were monitored on an Octet RED96. F10 was used as a nonbinding control.
To investigate whether these Abs can block virus attachment to cells, we next examined whether scFvFcs can inhibit S protein (Fig. S5A) or S-VLP (Fig. S5B) binding to hDPP4-expressing cells. As shown in Fig. S5A, all scFvFcs (except for 3B11, which gave maximal inhibition at all tested concentrations) could inhibit 50 nM S1(349–590)-hFc binding to 293T-hDPP4 cells in a dose-dependent manner. The control scFvFc, F10, did not inhibit S1(349–590)-hFc binding to 293T-hDPP4. In addition, Fig. S5B shows that all scFvFcs could inhibit the attachment of MERS S pseudovirus to hDPP4-expressing cells.
Generation of Ab Escape Mutants.
To investigate virus evolution under nAb pressure, we performed escape studies for all seven scFvFcs and three full-length IgG1s. Upon three rounds of selective pressure in the presence of each Ab, escape mutants for all the scFvFcs and for two of three IgG Abs were generated; no escape mutant was isolated for 3B11 IgG.
To map the mutations in the S protein that conferred escape to each Ab, all escape mutants were plaque purified, and the S protein-coding regions were sequenced. Only one or two amino acid substitutions conferred escape to each Ab (Fig. 4A). Interestingly, all the escape mutations mapped to the patch 2 region in the receptor-binding motif (RBM; amino acids 484–567) that interfaces with the hDPP4 (Fig. 4B), except for the escape mutant of 1F8 IgG1, which also had a distant substitution (I1229 T) in the S2 domain. Importantly, these escape mutations mirrored the Ab competition studies (Fig. S3) and confirmed the presence of three epitopes. Ab 1F8, 3A1, and 3B12 bind to amino acids in the center of the RBM, whereas 1E9 binds to epitope 1, and 3B11, 3C12, and M14D3 bind to another flanking epitope, epitope 3.
Fig. 4.

Escape mutations. (A) Amino acid differences among the RBDs of the S proteins of natural isolates and escape mutants (Em). (B) Diagram of the escape mutations in the RBD. The structure was adapted from Protein Dana Bank (PDB) ID code 4KR0. The RBM (amino acids 484–567) is colored in purple. The receptor-binding core is colored in light blue. The hDPP4 is colored gray. Three proposed epitopes are depicted as solid circles: epitope 1 in cyan, epitope 2 in magenta, and epitope 3 in green. Escape mutation residues are colored based on their corresponding epitope locations. Two major binding patches are shown by dotted circles shaded light green (patch 1) and orange (patch 2).
Escape Mutations Affect Abs’ Binding and Cross-Neutralization.
To investigate the affinities of the anti-S1 Abs binding to RBD mutants, seven RBD mutants were constructed, purified, and subjected to Octet analysis. As shown in Table S2, all seven Abs could bind to the L506F mutant with 0.52- to 7.7-fold affinity changes compared with their binding to the wild-type RBD. There also was a modest change of ∼0.97- to 5.26-fold binding affinity of all Abs to the P547S mutant. In addition, with the exception of 1E9 (epitope 1 Ab), binding affinity for all Abs to the other five RBD mutants carrying T512A, Y540C, or R542G mutations fell below the level of Octet detection. Interestingly, 1E9 maintains good binding affinity with both the Y540C mutant and the L506F and Y540C mutants, supporting the idea that1E9 binds to a distinct epitope, epitope 1, that does not overlap with epitope 3.
Additional PRNT50 assays were performed to assess whether higher concentrations of the selecting Ab could neutralize its escape virus (Fig. S6). All the escape mutants selected by these nAbs clearly escaped from their parent Abs, and eight of the escape mutants had IC50 values of ≥20 μg/mL Only Em3B11-Fc achieved an IC50 value of 6.76 μg/mL, a 3.7-fold increase compared with wild-type virus (Table S1).
Based on the epitope-mapping studies and locations of the escape amino acid changes, mutant viruses were assigned to three epitope groups. Cross-neutralization (PRNT50) assays were performed to analyze whether Abs of one group could still neutralize escape mutant viruses generated by Abs of another epitope group. IC50 values (Table S3) indicated that 1E9-Fc (group 1 Ab) could still neutralize escape viruses generated from group 2 (Em1F8-Fc) and group 3 (Em3B11-Fc, Em3C12-Fc) Abs; 1F8-Fc (group 2 Ab) could neutralize escape viruses generated from the group 1 (Em1E9-Fc) Ab and the group 3 [Em3B11-Fc (Y540C and L506F)] Ab with IC50 = 11.2 µg/mL, but not another group 3 [Em3C12-Fc (Y540H)]Ab with IC50 >40 µg/mL. Additionally, 3B11-Fc and 3C12-Fc (group 3 Abs) still could neutralize escape viruses from group 1 (Em1E9-Fc) and group 2 (Em1F8-Fc) Abs, although 3C12-Fc neutralizes Em1F8-Fc with an approximately sixfold higher IC50 (11.7 µg/mL). As expected, 1F8-Fc (a group 2 Ab) did not neutralize Em3A1-Fc (a group 2 escape virus) (IC50 >40 μg/mL). The 3B11-Fc (a group 3 Ab) could neutralize Em3C12-Fc (a group 3 escape virus) at a higher Ab concentration (IC50 = 8.94 vs. 1.83 for wild type), and 3C12-Fc (a group 3 Ab) could neutralize Em3B11-Fc (a group 3 escape virus) at a much higher Ab concentration (IC50 = 13.8 vs. 2.0 for wild type) (Table S3). These results indicate that a majority of these escape viruses generated from one Ab epitope group confer resistance to neutralization within the same Ab epitope group but not to a different Ab epitope group.
Effects of Escape Mutations on Viral Fitness.
To investigate how the escape mutations affect RBD–hDPP4 interaction and viral fitness, the binding affinities of seven mutated RBDs to hDPP4 were analyzed. As shown in Table S2, L506F and P547S had minimal effects on RBD–hDPP4 affinity, whereas the other three mutations (T512A, Y540C, and R542G) resulted in undetectable binding between the RBD mutants and hDPP4. To investigate these findings further, the RBD mutants were analyzed for hDPP4 binding by FACS, a more sensitive measure that may detect low-affinity binding. As shown in Fig. 5A, all seven mutated RBDs can bind to 293T-hDPP4 cells with varying efficacy. The P547S mutation did not change RBD binding to hDPP4 cells. However, the L506F single substitution caused an approximately threefold higher 50% effective concentration (EC50) than the wild-type RBD, whereas the other five mutants resulted in 10- to 25-fold higher EC50 values compared with the wild-type RBD.
Fig. 5.
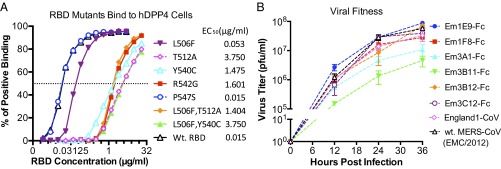
Characterization of RBD mutants. (A) Analysis of EC50 for the RBD mutant proteins binding to 293T-hDPP4 cells. Each purified RBD mutant, constructed with an hFc tag, was serially diluted 2× (12 dilutions starting from 5 or 20 µg/mL) and then was incubated with 1 × 105 293T-hDPP4. The binding of the RBD mutant proteins was monitored with APC-Cy7–labeled anti-hFc Abs by FACS. (B) In vitro growth kinetics of nAb escape mutant MERS-CoVs. Vero cells were infected with 0.1 MOI of escape mutant or wild-type MERS-CoV for 1 h in triplicate. Then unbound viruses were removed, growth medium was replenished, culture supernatants were sampled in triplicate at indicated time points, and virus titers were determined by plaque assay on Vero cells.
Six escape mutant viruses were chosen for further investigation of viral fitness (note that the Em1F8-IgG, Em3A1-IgG, and EmM14D3-Fc escape viruses were not evaluated because their substitutions are at R542, the same residue as in the chosen Em1F8-Fc and Em3A1-Fc escape viruses). All six neutralization escape viruses grew in Vero cells, reaching peak titers of 106–108 pfu/mL by 36 h after infection (Fig. 5B). Escape mutants Em3B11-Fc and Em3A1-Fc had 1- to 2-log reductions in titer at 12–36 h after infection, compared with the wild-type virus. Em1F8-Fc, Em3B12-Fc, and Em3C12-Fc had two- to fivefold reductions in titer, and Em1E9-Fc had a slightly increased viral titer. These results suggest that, by kinetic and peak titer assessment, most of the escape mutations resulted in impaired fitness.
We next compared the contact changes between the RBD and hDPP4 before and after neutralization escape to determine the potential atomic causes of changes in binding affinity. L506, W553, and V555 in the RBD and L294 and I295 in hDPP4 form a hydrophobic core in patch 2 at the protein–protein interface (23). As shown in Fig. S7 A and B and Table S4, the L506F substitution is predicted to result in a net decrease of four van der Walls (VDW) contacts between the RBD and hDPP4; however, the conserved hydrophobic L506F mutation may not change this hydrophobic core appreciably, in contrast to the significant loss of hDPP4 binding and cell-entry capacity with an L506A pseudovirus (23). The P547S substitution likely does not change the interaction of the RBD and hDPP4, because it is not in direct contact with hDPP4. In addition, although the orientation of the T512 side chain is not directly in contact with hDPP4, it makes 20 internal contacts within a loop structure that connects β-strands 6 and 7 (22). The T512 side chain does form an important hydrogen bond with D509 that may stabilize the loop structure (Fig. S7C). This hydrogen bond is lost with the T512A substitution, along with a 25% loss of other internal loop contacts (Fig. S7D). These atomic changes may alter the local conformation of the RBD, resulting in a decrease in hDPP4 binding (Table S4). In addition, although Y540 and R542 on the RBD can form hydrogen bonds with T265 and L294 on hDPP4, respectively (Fig. S7A), the Y540C substitution results in a shorter side chain, and the R542G substitution removes the side chain of this residue. These two substitutions may change the RBD–hDPP4 interaction dramatically, as illustrated in Fig. S7 A and B and Table S4, where Y540C and R542G substitutions resulted in a net of four and 11 decreases in VDW contacts between the RBD and hDPP4, respectively.
Discussion
In this study, we isolated seven anti–MERS-CoV Spike nAbs from a nonimmune human Ab-phage library using a novel panning strategy. These nAbs bind to three different epitopes in the RBD–hDPP4 interface with subnanomolar to nanomolar affinity and neutralize MERS-CoV infection. At least one major mechanism of action is their capacity to inhibit S protein binding to its DPP4 receptor. The residues within the RBD critical for neutralization were identified through the generation of escape variants and were confirmed by kinetic binding studies. Despite the close proximity of the three epitopes on the RBD interface, escape from one epitope minimally altered other Ab neutralization directed to a different epitope. Importantly, the majority of escape mutations had negative impacts on hDPP4 receptor binding and viral fitness. These results provide insight into the basis and biases of human nAbs against MERS-CoV that may contribute to MERS-CoV clearance and evolution.
We used mammalian cell-produced full-length S protein expressed on PMPLs and cell surfaces for Ab panning. Surprisingly, all Abs identified were directed to the RBD and have potent neutralizing activity. With this same Ab-phage library, we previously had identified potent neutralizing Abs against SARS-S and broadly cross-neutralizing Abs against the fusion peptide of West Nile virus E protein and the hemagglutinin stem domain of influenza A viruses (29, 34, 35, 37). In contrast to our previous report on the isolation of RBD-directed nAbs against SARS-CoV using immunotube-bound S1 protein as bait (35), the combined strategy of sequentially using full-length S protein expressed on PMPLs and then on cells as bait resulted in a higher recovery of potent nAbs and further demonstrates that the RBD interface is dominant in our selection system. The presentation of the native S glycoprotein as an intact trimer on PMPLs and cells may present the S1 domain more effectively than the S protein immobilized on immunotubes. Thus, this technology again has proven valuable for obtaining potent human nAbs in the absence of specimens from immune patients containing plasmablasts or memory B cells (31, 38). This distinction is particularly important when early access to patient specimens is problematic, in part because of government, regulatory, and biocontainment restrictions. Thus, this rapid response platform is well positioned to isolate and characterize robust immunotherapeutics against future emerging viruses quickly.
Abs typically accumulate 5–20% amino acid changes in their V-segments during the affinity maturation process, which consist of 6–25 amino acid changes in the VH or VL domains (39). NAbs against HIV gp120 often have amino acid changes of 19–46% in their VH genes alone (40). The seven anti-Spike nAbs described here use three VH and six VL germ-line genes with 0–10 SHMs. Interestingly, five of seven Abs use the IGHV1-69 germ-line gene that is preferentially used by many anti-viral nAbs, including those directed against hepatitis C virus and influenza viruses (34, 39, 41). IGHV1-69 is the only VH gene that has two hydrophobic residues at the tip of its CDR–H2 loop that can make direct contact with the viral glycoprotein (42). The Abs 3B11 and 3B12 use the IGHV1-69*06 germ-line gene without any SHMs and have only nine or three SHMs in their light chains, respectively, but they maintain subnanomolar binding affinities for S. The hydrophobic core of patch 2 at the RBD–hDPP4 interface is in the center of these epitopes. We suggest that the IGHV1-69 Abs may replace hDPP4 and make direct contact with this hydrophobic core through their CDR–H2 loops (23). Our finding that no to only a few SHMs are required for RBD binding further suggests that a potentially greater number of IGHV1-69 B-cell receptor precursors may be able to recognize the RBD and that a prolonged affinity maturation process is not required to develop a potent nAb response. In addition, there are 14 IGHV1-69 alleles and significant copy number variation (CNV), from zero to five copies, because of gene deletion and duplication (43, 44). The IGHV3-30 and IGHV1-3 germ-line genes used by 1E9 and 1F8, respectively, also have multiple alleles and have been reported for other anti-viral Abs (39, 45). Whether this Ig polymorphism and CNV can influence the early nAb response to and the clinical outcome of MERS-CoV infection is not known. The limited clinical data that are available have reported only that most MERS patients have had immunosuppression and/or underlying disease (46, 47).
Human DPP4 binds to the MERS-CoV RBD with a Kd of ∼21.4 nM, which is comparable with a previous report 16.7 nM (22). All seven nAbs bind to the RBD with 10- to 450-fold higher affinity than to hDPP4. Previous studies have indicated that higher Ab affinity confers broader and higher neutralizing activity (48). Here, we dissect the contributions of binding affinity and epitope specificity toward defining nAb activities. When binding to the same epitope, nAbs with higher binding affinity had higher neutralizing activity. Among the epitope group 2 Abs, 3A1 scFvFc had a higher affinity than 1F8 and exhibited better neutralization activity (Fig. 2A and Table 1). Among the epitope group 3 Abs, 3B11 scFvFc/IgG had a higher affinity and showed better neutralizing activity than 3C12 scFvFc/IgG (Fig. 2 A and B and Table 1). However, when nAbs bind to different epitopes, the nAbs binding to the center of the RBD–hDPP4 interface are more potent. For example, the epitope group 1 Ab 1E9 has strong binding affinity to the RBD but has weak neutralization activity (Fig. 2A and Table 1), perhaps because 1E9 binds to the small η4 helix that is close to but distinct from the RBD–hDPP4 interface. Binding to this position may not completely block hDPP4 binding to the RBD. In contrast, 3C12 has lower binding affinity than 1E9 but has better neutralization activity because it binds directly at the RBD–hDPP4 interface (Fig. 4B).
The generation of neutralization escape mutants can be a helpful tool for identifying residues critical for neutralization and for investigating virus evolution under immune pressure (37, 49). Like other RNA viruses, CoVs have high mutation rates, especially during cross-species transmission, that are important for virus adaptation to new host receptors (5, 6, 50). Immune pressure is another force selecting for virus mutation (37, 49). With the exception of the 3B11 IgG, which appears to have a binding affinity below the neutralization escape threshold (Table 1), all other Abs, including 3B11 scFvFc, underwent neutralization escape. Based on Ab studies against a number of viral pathogens by ourselves and other investigators, we assume that the escape mutants from these nAbs may reflect virus evolution under similar nAb-mediated immune pressure in patients. In vitro escape studies showed that a single-amino acid mutation at residues Y540 and R542 could confer virus neutralization escape from four nAbs (1F8, 3A1, 3C12, and M14D3). For the 3B11 and 3B12 Abs, which use the germ-line gene of VH1-69*06, two escape mutations were observed for each nAb, although further investigation indicated that the L506F substitution alone could not abrogate 3B11 and 3B12 nAb binding; in contrast, the respective Y540C and T512A substitutions resulted in the profound loss of nAb binding activity. Natural virus variation with F506 has been reported for England-1 MERS-CoV, which was isolated from the second MERS patient from Qatar (51). It is possible that 3B11- or 3B12-like nAbs were involved in driving the L506F mutation (52). We tested the neutralization of 3B11 and 3B12 scFvFcs on the England-1 CoV strain, which encodes the L506F substitution in the S protein. The respective IC50 values for 3B11-Fc and 3B12-Fc were 5.48 and 13.8 µg/mL, demonstrating that three- or 11-fold higher concentrations were required to neutralize the England-1 MERS-CoV strain compared with the EMC/2012 MERS-CoV strain.
The effects of these escape mutations on hDPP4 binding activity and viral fitness also are significant. Both BLI and FACS assessments of RBD mutants indicated that, except for the P547S substitution that was selected by the epitope 1 Ab 1E9, all other substitutions exhibited either small (L506F) or significantly (T512A, Y540C, R542G) decreases in RBD binding affinity to hDPP4 (Table S2). Viral fitness assays demonstrated that the P547S substitution slightly enhanced viral fitness, whereas the substitutions at positions T512, Y540, and R542 of epitopes 2 and 3 decreased viral fitness. Although we did not generate a viral escape mutant carrying the L506F substitution alone, the decrease in binding affinity for hDPP4 caused by the L506F substitution may contribute to its decreased viral fitness (25) (Fig. 5A and Table S2). These results lead us to propose that the immunodominance of the human nAb response to the RBD–hDPP4 interface can restrict MERS-CoV evolution by driving virus down an escape pathway that predominantly results in a significant cost in viral fitness. In vitro human mAb neutralization studies on SARS-CoV recapitulated the escape mutations that were seen during the SARS outbreak (37). Additionally, decreased virulence of the neutralization escape mutants coincided with reduced affinity of the mutant SARS-S for its ACE2 receptor (49). Given the decreased growth kinetics of these escaped mutants in vitro, we anticipate that the majority of escape mutants selected from similar nAbs in vivo may be attenuated.
In summary, this study identified potent anti–MERS-CoV S nAbs that bind to three epitopes at the RBD interface and show minimal evidence of cross-epitope resistance. We suggest that MERS-CoV vaccines, particularly those that express S in the context of virus/VLP membranes, as we describe here, may be an effective way to present this vulnerable interface for immune recognition (26, 27). In addition, in view of current recommendations by the International Severe Acute Respiratory and Emerging Infection Consortium and the increasing recognition that human Abs may have a role in the management of infectious diseases, the therapeutic potential of these nAbs should be considered for the prophylaxis and treatment of MERS (53). Although escape from neutralization is a concern with therapeutic Abs, our study provides reagents and a strategy to mitigate this potential problem. Divergent combination immunotherapy uses two different nAbs to non–cross-resistant epitopes to decrease the possibility of viral escape while taking advantage of synergistic neutralizing effects (37, 49, 54, 55). By virtue of its high affinity and lack of neutralization escape, the 3B11 IgG should be included in this mAb mixture. In addition, our immunogenetic results provide the basis for comparisons with more global antibodyome studies of circulating B cells from MERS-CoV–infected patients during acute and/or convalescent phases of infection and an opportunity to examine the role that Ig polymorphism may play in shaping the protective Ab repertoire and influencing clinical responses (44, 56).
Materials and Methods
Expression and Purification of the MERS-CoV Spike.
The MERS-CoV S gene (according to GenBank accession no. JX869059) was codon-optimized and synthesized for mammalian cell expression. The full-length Spike with a Flag tag (DYKDDDDK) at its N terminus (between the signal peptide and the extracellular domain of the S protein) and a C9 tag (TETSQVAPA) at its C terminus was first cloned into the pcDNA3.1 vector. Subsequently a series of constructs encoding different fragments of MERS-CoV Spike protein was designed. Plasmids encoding MERS-CoV S protein residues 21–751, 752–1295, 21–358, 349–751, and 349–590 fused with the CD5 signal peptide and the constant region fragment of human IgG1 were named S1-hFc, S2-hFc, S1(21–358)-hFc, S1(349–571)-hFc, and S1(349–590)-hFc (RBD-hFc), respectively (Fig. S1). The last-named three constructs also have Flag tags at their N termini. Fc-tagged proteins were expressed from transiently transfected 293T cells and were purified with protein A Sepharose (GE Healthcare).
Stable 293T Cells Expressing MERS-CoV Spike or Human DPP4.
Full-length S with an N-terminal Flag tag and a C-terminal C9 tag was cloned into the pHAGE lentiviral vector, which can express both S and ZsGreen via an internal ribosome entry site sequence. Corresponding lentiviral particles pseudotyped with Vesicular stomatitis virus-G (VSV-G) envelope proteins were produced from Lenti-X 293T cells as described and were used for 293T cell transduction (57). The cells expressing the MERS S protein then were sorted by flow cytometry with GFP. After 2–3 wk of propagation, stable S-293T cells were sorted again with anti-Flag Ab and set aside for future experiments. hDPP4 was RT-PCR–amplified from human peripheral blood mononuclear cells and then was cloned into a pHAGE vector without a ZsGreen reporter gene. Stable 293T-hDPP4 cells were established following a protocol similar to the one described above.
Preparation of MERS Spike-PMPLs.
The S-PMPLs were prepared as described elsewhere (58, 59). Briefly, 1 × 108 S-293T cells were lysed with lysis buffer and then were incubated with 5 × 108 M-280 Tosylactivated Dynabeads (Dynal Biotech) coated with C9 tag mAb (1D4). The Spike-bound beads were washed extensively and resuspended in 1.25 mL buffer containing 100 mM (NH4)2SO4, 20 mM Tris (pH 7.5), 10% (vol/vol) glycerol, and 1 mg/mL lipid mixture consisting of 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine/1,2-Dioleoyl-sn-glycero-3-phosphate (POPC/POPE/DOPA) (6:3:1; Avanti Polar Lipids). The protein-liposome mixture was dialyzed to remove the detergent slowly and to allow the formation of proteoliposomes. The dialyzed beads were washed twice with 0.1% BSA/PBS and then were resuspended in 0.1%BSA/PBS to a concentration of 1 × 109/mL. The protein composition of S-PMPL and of control 293T-PMPL cells then was analyzed by SDS/PAGE and Western blot.
Selection of Phage Abs by Panning with S-PMPLs and S-293T Cells.
Two combined human scFv libraries (Mehta I/II) (∼27 billion members) constructed from B cells of 57 unimmunized donors were mixed and used for selection of scFvs against MERS-CoV S. 293T cells, 293T-PMPLs, and S-PMPLs were preblocked in 2% BSA/2% nonfat milk/PBS for 1 h at room temperature. An aliquot of the mixed phage library (5 × 1012 pfu) was incubated with 3 × 107 293T cells and 3 × 107 293T-PMPLs three times to remove the nonspecific phage clones. Preabsorbed phage was incubated with 2 × 107 S-PMPLs in 2 mL of 2% BSA/2% nonfat milk/PBS for 2 h at room temperature with gentle shaking. Nonspecifically absorbed phages were removed by intensive washing with PBS containing 0.1% Tween 20. Specifically bound phages were eluted with 100 mM triethylamine and then were neutralized, amplified, and used for the next round of panning. After two rounds of panning with S-PMPL, the eluted phages were incubated directly with S-293T cells, followed by PerCP-Cy5.5–labeled anti-M13 Ab staining. PerCP-Cy5.5 and GFP double-positive cells were sorted by FACS and used to infect TG1 cells. Randomly picked single phage-scFv clones were screened for specific binding to S1-hFc and S2-hFc by MSD and ELISA as previously described (35, 59). The VH and VL of MSD/ELISA-positive clones were sequenced and aligned for the identification of unique Ab clones.
Expression and Purification of scFvFcs and Whole Human IgG1.
The VH and VL scFvs of unique positive clones were first cloned into a eukaryotic expression vector, where the scFv is fused in-frame with the human IgG1 hinge-CH2-CH3 domains. For production of whole human IgG1, the VH and VL gene fragments of scFv were subcloned separately into the human IgG1κ expression vector TCAE5 or the IgG1λ expression vector TCAE6. These scFvFcs/IgG1s were transiently transfected into 293F cells, and the cell culture supernatants were collected twice every 72 h. The expressed scFvFcs and IgG1 were purified by protein A Sepharose affinity chromatography (GE Healthcare).
S1 Domain Epitope Mapping and Binding Competition.
Recombinant soluble S1(21–358)-hFc, S1(349–751)-hFc, and S1(349–590)-hFc were immobilized to anti-Flag biosensor at 30 °C for 240 s. The association of 100 nM scFvFcs 1E9, 1F8, 3A1, 3B11, 3B12, 3C12, and M14D3 with each Spike was measured by using an Octet RED96 (ForteBio, Inc.) for 240 s at 30 °C by exposing the sensors to 100 nM Ab in 1× kinetic buffer. ScFvFc of F10 was used as a nonbinding control.
For the binding competition assay, recombinant soluble S1(349–590)-hFc was bound to an anti-Flag biosensor. The association of each scFvFcs was measured on the Octet RED96 for 300 s at 30 °C by exposing the sensors to 100 nM scFvFc in 1× kinetic buffer; then the degree of additional binding was assessed by exposing the sensors to a second scFvFc (100 nM in 1× kinetic buffer) in the presence of the first scFvFc (100 nM) for 300 s at 30 °C. F10 was used as a nonbinding control.
Affinity Measurement by Octet.
Ab binding affinity was determined using the same Octet RED96 instrument. Purified S1(349–590)-hFc at 5 μg/mL in 1× kinetic buffer was immobilized onto anti-Flag biosensors and incubated with varying concentrations of Abs in solution. All binding data were collected at 30 °C. The experiments included five steps: (i) baseline (60 s); (ii) S1(349–590)-hFc loading onto sensors (300 s); (iii) second baseline (120 s); (iv) association of Abs for measurement of Kon (300 s); and (v) dissociation of Abs for the measurement of Koff (600–1,200 s). Baseline and dissociation steps were conducted in 1× kinetic buffer. Fitting curves were constructed using ForteBio Data Analysis 7.0 software.
Neutralization Assay with MERS-CoV Spike Pseudotyped Lentivirus.
MERS-VLPs were produced by cotransfecting Lenti-X 293T cells with pcDNA3.1-Sf (encoding the full-length S of MERS-CoV), pHIV-Luc (encoding the luciferase reporter gene), and pCMV-ΔR8.2 (encoding HIV gag-pol). Fivefold serially diluted Abs beginning at 10 μg/mL were incubated in a 96-well plate with an equal volume of pseudovirus at the final volume of 60 μL at room temperature for 1 h, and the mixture was added to the monolayer of 293T-hDPP4 cells cultured in a luminometer plate (PerkinElmer). Anti-influenza HA F10 Ab (scFvFc and IgG1) was used as the control Ab. VSV-G pseudotyped lentivirus was used as the virus control. After overnight incubation, the plate was replenished with 100 μL fresh medium in each well and cultured for another 24 h. Luciferase activity was measured by using a luciferase assay kit (Promega). Briefly, culture medium was removed, cells were lysed by the addition of 30 μL lysis buffer, and the relative luciferase units in cell lysates were measured using a POLARstar Omega (BMG Labtech). This assay was performed in triplicate.
Plaque Reduction Neutralization Test with Live MERS-CoV.
MERS-CoV EMC/2012 was cultured on Vero cells. The neutralization assay was conducted using a PRNT50 assay as described previously (48, 60). Briefly, serial dilutions of the Abs in PBS were incubated with 100 pfu of MERS-CoV for 1 h at 37 °C, and the Ab-virus mix was added to the monolayer of Vero cells, along with virus without Ab. After 1 h of incubation, overlay-containing medium and agarose were added, and the plaques formed in each dilution were counted 48–72 h following infection. The percentage of neutralization was calculated as [(100 − number of plaques with Ab)/number of plaques without Ab)] × 100.
Binding Competition Between scFvFcs and DPP4.
Recombinant soluble human DPP4 was purchased from Abcam. A buffer exchange step to PBS was performed using an Amicon Ultra 0.5-mL centrifugal filter (Millipore). Purified S1(349–590)-hFc was loaded onto anti-Flag biosensors and incubated first with 100 nM of each scFvFcs. Additional binding of DPP4 was measured by exposing the sensors to 100 nM DPP4 in the presence of the 100 nM scFvFc. Similarly, anti-Flag biosensors loaded with S1(349–590)-hFc was incubated with 100 nM of DPP4 followed by monitoring the subsequent binding of 100 nM scFvFc to the sensors in the presence of 100 nM DPP4. All measurements were performed on an Octet RED96.
Ab Inhibition of Spike and MERS-VLP Binding to 293T-hDPP4 Cells.
S1(349–590)-hFc (50 nM) was incubated with scFvFcs (450, 150, 50, or 0 nM) in a 50-μL volume at 4 °C for 1 h. F10 scFvFc was used as the control. Each mixture was added to 1 × 105 293T-hDPP4 cells and incubated at 4 °C for 1 h. Cells were washed three times with PBS containing 0.5% BSA and 0.1% NaN3. For detection of S1(349–590)-hFc binding to 293T-hDPP4 cells, cells were incubated with mouse anti-Flag Ab at 4 °C for 30 min followed by APC-Cy7–labeled goat anti-mouse IgG (Pierce). After extensive washing, samples were analyzed using a BD LSRII.
Similarly, 50 μL MERS-VLP was mixed with scFvFcs (450, 150, 50, or 0 nM) at 4 °C for 1 h. The mixtures then were added separately to 293T-hDPP4 cells. After washing, the binding of MERS-VLPs to 293T-hDPP4 cells was monitored with anti-Flag Ab staining and flow cytometry.
Isolation of the Escape Mutants Under Selective Pressure of nAbs.
MERS-CoV (1 × 106 pfu) was incubated with 30 µg of each Ab in a 200-µL volume of PBS for 30 min and then was added to a monolayer of Vero cells. The development of cytopathic effect (CPE) was monitored over 72 h, and progeny viruses were harvested. Three rounds of selections were performed in the presence of 30 µg/mL Abs (except for 3B11 scFvFc), and CPE was noted during each passage. The viruses from final passage were plaque purified in the presence of Ab, and titers were determined as described (48). The S genes from plaque-purified viruses were sequenced to identify mutations that conferred escape from these Abs.
Escape Mutations Affect RBD-Ab Binding and Cross-Neutralization.
Seven RBD mutant derivatives of S1(349–590)-hFc were constructed (Table S2) using a site-directed mutagenesis kit (Agilent). The mutant RBD proteins were expressed and purified from transfected 293T cells. The binding affinities of seven scFvFcs to these RBD mutants were measured on an Octet RED96 as described above. Furthermore, Ab neutralization against escape mutants was investigated by PRNT50 assay.
Escape Mutations Affect RBD-hDPP4 Binding and Viral Fitness.
The binding affinity of RBD mutants to hDPP4 was measured first using the Octet RED96. Because five of seven RBD mutants have undetectable binding to hDPP4, the RBD-hDPP4 binding was investigated further by FACS. Each RBD mutants was serially diluted 2× and incubated with 1 × 105 293T-hDPP4 cells in 100 µL of FACS buffer. After three washings, cells were stained with APC-Cy7–labeled goat anti-human IgG1. After extensive washing, the samples were analyzed by using a BD LSRII.
Virus growth kinetics were measured in triplicate by inoculating Vero cells with different escape mutants at a multiplicity of infection (MOI) of 0.1 for 1 h, after which unbound virus was washed, medium was added back, and the supernatant was sampled at various time points. Virus titers were determined by plaque assay.
Supplementary Material
Acknowledgments
We thank Drs. Zhen Zhang and De-Kuan Chang for helpful discussions and Dr. Maria Zambon and Dr. Robin Gopal at Public Health England for providing the MERS-CoV England-1 strain. This work was supported by Defense Advanced Research Projects Agency Grant W911NF-10-0226 (to W.A.M.) and National Institutes of Health Grant R01AI085524 (to W.A.M. and R.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1402074111/-/DCSupplemental.
References
- 1.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 2.Breban R, Riou J, Fontanet A. Interhuman transmissibility of Middle East respiratory syndrome coronavirus: Estimation of pandemic risk. Lancet. 2013;382(9893):694–699. doi: 10.1016/S0140-6736(13)61492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Health Protection Agency (HPA) UK Novel Coronavirus Investigation team Evidence of person-to-person transmission within a family cluster of novel coronavirus infections, United Kingdom, February 2013. Euro Surveill. 2013;18(11):20427. doi: 10.2807/ese.18.11.20427-en. [DOI] [PubMed] [Google Scholar]
- 4.Cotten M, et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. MBio. 2014;5(1):e01062–e01013. doi: 10.1128/mBio.01062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chinese SARS Molecular Epidemiology Consortium Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303(5664):1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 7.Annan A, et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg Infect Dis. 2013;19(3):456–459. doi: 10.3201/eid1903.121503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anthony SJ, et al. Coronaviruses in bats from Mexico. J Gen Virol. 2013;94(Pt 5):1028–1038. doi: 10.1099/vir.0.049759-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo PC, et al. Molecular diversity of coronaviruses in bats. Virology. 2006;351(1):180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Memish ZA, et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg Infect Dis. 2013;19(11):1819–1823. doi: 10.3201/eid1911.131172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perera RA, et al. Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt, June 2013. Euro Surveill. 2013;18(36):20574. doi: 10.2807/1560-7917.es2013.18.36.20574. [DOI] [PubMed] [Google Scholar]
- 12.Reusken CB, et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: A comparative serological study. Lancet Infect Dis. 2013;13(10):859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haagmans BL, et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect Dis. 2014;14(2):140–145. doi: 10.1016/S1473-3099(13)70690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer B, et al. Antibodies against MERS Coronavirus in Dromedary Camels, United Arab Emirates, 2003 and 2013. Emerg Infect Dis. 2014;20(4):552–559. doi: 10.3201/eid2004.131746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alagaili AN, et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in saudi arabia. MBio. 2014;5(2):e00884–e00814. doi: 10.1128/mBio.00884-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckerle I, et al. Replicative Capacity of MERS Coronavirus in Livestock Cell Lines. Emerg Infect Dis. 2014;20(2):276–279. doi: 10.3201/eid2002.131182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller MA, et al. Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. MBio. 2012;3(6):e00515–e00512. doi: 10.1128/mBio.00515-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofmann H, et al. S protein of severe acute respiratory syndrome-associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J Virol. 2004;78(12):6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan KH, et al. Cross-reactive antibodies in convalescent SARS patients’ sera against the emerging novel human coronavirus EMC (2012) by both immunofluorescent and neutralizing antibody tests. J Infect. 2013;67(2):130–140. doi: 10.1016/j.jinf.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gierer S, et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol. 2013;87(10):5502–5511. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raj VS, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495(7440):251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu G, et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature. 2013;500(7461):227–231. doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang N, et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013;23(8):986–993. doi: 10.1038/cr.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falzarano D, et al. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat Med. 2013;19(10):1313–1317. doi: 10.1038/nm.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agnihothram S, et al. Evaluation of serologic and antigenic relationships between middle eastern respiratory syndrome coronavirus and other coronaviruses to develop vaccine platforms for the rapid response to emerging coronaviruses. J Infect Dis. 2014;209(7):995–1006. doi: 10.1093/infdis/jit609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du L, et al. A truncated receptor-binding domain of MERS-CoV spike protein potently inhibits MERS-CoV infection and induces strong neutralizing antibody responses: Implication for developing therapeutics and vaccines. PLoS ONE. 2013;8(12):e81587. doi: 10.1371/journal.pone.0081587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song F, et al. Middle East respiratory syndrome coronavirus spike protein delivered by modified vaccinia virus Ankara efficiently induces virus-neutralizing antibodies. J Virol. 2013;87(21):11950–11954. doi: 10.1128/JVI.01672-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marasco WA, Sui J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol. 2007;25(12):1421–1434. doi: 10.1038/nbt1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gould LH, et al. Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J Virol. 2005;79(23):14606–14613. doi: 10.1128/JVI.79.23.14606-14613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kashyap AK, et al. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc Natl Acad Sci USA. 2008;105(16):5986–5991. doi: 10.1073/pnas.0801367105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Traggiai E, et al. An efficient method to make human monoclonal antibodies from memory B cells: Potent neutralization of SARS coronavirus. Nat Med. 2004;10(8):871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333(6044):850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 33.Throsby M, et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE. 2008;3(12):e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sui J, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16(3):265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sui J, et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc Natl Acad Sci USA. 2004;101(8):2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mou H, et al. The receptor binding domain of the new Middle East respiratory syndrome coronavirus maps to a 231-residue region in the spike protein that efficiently elicits neutralizing antibodies. J Virol. 2013;87(16):9379–9383. doi: 10.1128/JVI.01277-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sui J, et al. Broadening of neutralization activity to directly block a dominant antibody-driven SARS-coronavirus evolution pathway. PLoS Pathog. 2008;4(11):e1000197. doi: 10.1371/journal.ppat.1000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wrammert J, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453(7195):667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013;31:705–742. doi: 10.1146/annurev-immunol-032712-095916. [DOI] [PubMed] [Google Scholar]
- 40.Wu X, et al. NISC Comparative Sequencing Program Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333(6049):1593–1602. doi: 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan CH, Hadlock KG, Foung SK, Levy S. V(H)1-69 gene is preferentially used by hepatitis C virus-associated B cell lymphomas and by normal B cells responding to the E2 viral antigen. Blood. 2001;97(4):1023–1026. doi: 10.1182/blood.v97.4.1023. [DOI] [PubMed] [Google Scholar]
- 42.Huang CC, et al. Structural basis of tyrosine sulfation and VH-gene usage in antibodies that recognize the HIV type 1 coreceptor-binding site on gp120. Proc Natl Acad Sci USA. 2004;101(9):2706–2711. doi: 10.1073/pnas.0308527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyd SD, et al. Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J Immunol. 2010;184(12):6986–6992. doi: 10.4049/jimmunol.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watson CT, Breden F. The immunoglobulin heavy chain locus: Genetic variation, missing data, and implications for human disease. Genes Immun. 2012;13(5):363–373. doi: 10.1038/gene.2012.12. [DOI] [PubMed] [Google Scholar]
- 45.Farci P, et al. B cell gene signature with massive intrahepatic production of antibodies to hepatitis B core antigen in hepatitis B virus-associated acute liver failure. Proc Natl Acad Sci USA. 2010;107(19):8766–8771. doi: 10.1073/pnas.1003854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Assiri A, et al. KSA MERS-CoV Investigation Team Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med. 2013;369(5):407–416. doi: 10.1056/NEJMoa1306742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Memish ZA, Zumla AI, Al-Hakeem RF, Al-Rabeeah AA, Stephens GM. Family cluster of Middle East respiratory syndrome coronavirus infections. N Engl J Med. 2013;368(26):2487–2494. doi: 10.1056/NEJMoa1303729. [DOI] [PubMed] [Google Scholar]
- 48.Rani M, et al. Increased antibody affinity confers broad in vitro protection against escape mutants of severe acute respiratory syndrome coronavirus. J Virol. 2012;86(17):9113–9121. doi: 10.1128/JVI.00233-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rockx B, et al. Escape from human monoclonal antibody neutralization affects in vitro and in vivo fitness of severe acute respiratory syndrome coronavirus. J Infect Dis. 2010;201(6):946–955. doi: 10.1086/651022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cotten M, et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive genomic study. Lancet. 2013;382(9909):1993–2002. doi: 10.1016/S0140-6736(13)61887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bermingham A, et al. Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East, September 2012. Euro Surveill. 2012;17(40):20290. [PubMed] [Google Scholar]
- 52.Cotten M, et al. Full-genome deep sequencing and phylogenetic analysis of novel human betacoronavirus. Emerg Infect Dis. 2013;19(5):736–42B. doi: 10.3201/eid1905.130057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Public Health England, ISARIC (2013) Treatment of MERS-CoV: Decision Support Tool. Clinical Decision Making Tool for Treatment of MERS-CoV v.1.1. Available at www.hpa.org.uk/webc/HPAwebFile/HPAweb_C/1317139281416. Accessed April 11, 2014.
- 54.ter Meulen J, et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 2006;3(7):e237. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bakker AB, et al. First administration to humans of a monoclonal antibody cocktail against rabies virus: safety, tolerability, and neutralizing activity. Vaccine. 2008;26(47):5922–5927. doi: 10.1016/j.vaccine.2008.08.050. [DOI] [PubMed] [Google Scholar]
- 56.Dimitrov DS. Therapeutic antibodies, vaccines and antibodyomes. MAbs. 2010;2(3):347–356. doi: 10.4161/mabs.2.3.11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taube R, et al. Lentivirus display: stable expression of human antibodies on the surface of human cells and virus particles. PLoS ONE. 2008;3(9):e3181. doi: 10.1371/journal.pone.0003181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mirzabekov T, Kontos H, Farzan M, Marasco W, Sodroski J. Paramagnetic proteoliposomes containing a pure, native, and oriented seven-transmembrane segment protein, CCR5. Nat Biotechnol. 2000;18(6):649–654. doi: 10.1038/76501. [DOI] [PubMed] [Google Scholar]
- 59.Xu C, Sui J, Tao H, Zhu Q, Marasco WA. Human anti-CXCR4 antibodies undergo VH replacement, exhibit functional V-region sulfation, and define CXCR4 antigenic heterogeneity. J Immunol. 2007;179(4):2408–2418. doi: 10.4049/jimmunol.179.4.2408. [DOI] [PubMed] [Google Scholar]
- 60.Rockx B, et al. Structural basis for potent cross-neutralizing human monoclonal antibody protection against lethal human and zoonotic severe acute respiratory syndrome coronavirus challenge. J Virol. 2008;82(7):3220–3235. doi: 10.1128/JVI.02377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.