

Significance
HIV-1 enters host cells via CD4 and the coreceptors CXCR4 or CCR5. Most HIV-1 variants isolated from newly infected individuals use CCR5 (R5 strains) and infect Th1 cells, among other cell types. In ∼50% of patients, R5 strains shift to X4 strains (which use CXCR4) and infect mainly Th2 cells, leading to poor prognosis and rapid disease progression. In Th2 cells, CD4 and CXCR4 levels resemble those of Th1 cells, but they express little CCR5. We report that CCR5 expression in CD4+ T cells reduced X4 strain cell entry and infection; the molecular mechanism involves CD4/CXCR4/CCR5 oligomer formation. CCR5 expression altered CD4/CXCR4 heterodimer conformation, blocking virus binding. Oligomeric complexes should thus be considered a target for reducing HIV-1 binding and infection.
Keywords: chemokine receptors, oligomer formation, FRET/BRET
Abstract
CCR5 and CXCR4, the respective cell surface coreceptors of R5 and X4 HIV-1 strains, both form heterodimers with CD4, the principal HIV-1 receptor. Using several resonance energy transfer techniques, we determined that CD4, CXCR4, and CCR5 formed heterotrimers, and that CCR5 coexpression altered the conformation of both CXCR4/CXCR4 homodimers and CD4/CXCR4 heterodimers. As a result, binding of the HIV-1 envelope protein gp120IIIB to the CD4/CXCR4/CCR5 heterooligomer was negligible, and the gp120-induced cytoskeletal rearrangements necessary for HIV-1 entry were prevented. CCR5 reduced HIV-1 envelope-induced CD4/CXCR4-mediated cell-cell fusion. In nucleofected Jurkat CD4 cells and primary human CD4+ T cells, CCR5 expression led to a reduction in X4 HIV-1 infectivity. These findings can help to understand why X4 HIV-1 strains infection affect T-cell types differently during AIDS development and indicate that receptor oligomerization might be a target for previously unidentified therapeutic approaches for AIDS intervention.
For HIV-1 to enter a target cell, the viral envelope glycoprotein gp120 must interact with a set of cell surface molecules that include the primary receptor, CD4 (1), and a chemokine receptor (CCR5 or CXCR4) that acts as a coreceptor (2, 3). These molecules form CD4/chemokine receptor complexes, as deduced from coprecipitation data for CXCR4 or CCR5 with CD4 (4–8).
Most HIV-1 variants isolated from newly infected individuals use CCR5 and CD4 to enter host cells; these M-tropic R5 strains are predominant in acute and asymptomatic phases of HIV infection. CD4+ T helper type 1 (Th1) cells, which express high CCR5 levels (9, 10), are implicated in maintaining asymptomatic status (11, 12). The “viral shift” from R5 to T-tropic X4 HIV-1 strains correlates with AIDS progression (13, 14). X4 strains infect mainly CD4+ Th2 cells, which express little CCR5 and whose CXCR4 levels resemble those of Th1 cells (15, 16), which suggests that cell susceptibility to HIV-1 infection depends on the CD4/coreceptor ratio and on receptor levels during cell activation and/or differentiation (17). CXCR4 and CCR5 are present as homodimers and heterodimers at the plasma membrane (18–20). In addition, gp120-mediated CD4/CXCR4 and CD4/CCR5 association and clustering is reported (21–23). Nonetheless, little is known of how CCR5 expression influences the CD4/CXCR4 interaction, or of the molecular basis that underlies the differences in X4 strains infection relative to CCR5 levels at the cell surface.
Here, we identify CD4/CXCR4/CCR5 oligomers at the cell membrane, even in the absence of ligands. CCR5 expression in these complexes modifies the heterodimeric CD4/CXCR4 conformation and blocks gp120IIIB binding, without altering binding of the CXCR4 ligand CXCL12 and its subsequent signaling. gp120IIIB-triggered LIMK1 activation, cofilin dephosphorylation, and the actin cytoskeleton rearrangement necessary for cell-cell fusion were impeded in CD4/CXCR4/CCR5-expressing cells. The data obtained using recombinant gp120IIIB glycoprotein were confirmed by experiments showing that X4 HIV-1 infection of Jurkat and primary T cells is regulated by CCR5 expression.
Results
CD4, CXCR4, and CCR5 Form a Heterocomplex in Living Cells.
Chemokine receptors can form homodimers and heterodimers (18–20) (Fig. S1). Bioluminescence resonance energy transfer (BRET) titration assays were used to test CD4 heterodimeric complex formation with CXCR4 and CCR5. We cotransfected 293T cells with a constant amount of donor [CD4-Rluc (renilla luciferase)] and increasing amounts of acceptor (CXCR4-CFP or CCR5-YFP) and then analyzed in BRET2 or BRET1 assays, respectively. Fusion of the luciferase protein to the CD4 C-terminal tail did not alter receptor expression or function (Fig. S2 A and B). Using the Dako Cytomation Qifikit, we confirmed that CD4-Rluc–transfected 293T cells expressed the protein within the physiological range, i.e., similar to amounts in CD4+ primary T cells (293T cells, 13,828 ± 3,686 CD4 molecules per cell). BRET was positive for CD4/CXCR4 (BRET50 18.01 ± 10.08) and for CD4/CCR5 (BRET50 7.46 ± 2.63) (Fig. 1 A and B). These results are consistent with the constitutive association between CD4 and the coreceptors detected by coprecipitation in monocytes and macrophages (4–8).
Fig. 1.

CD4 forms heterodimers with CXCR4 and CCR5. (A) BRET2 experiment scheme of the postulated interaction between CD4-Rluc and CXCR4-CFP (Upper). We generated BRET titration curves by using 293T cells transiently cotransfected with CD4-Rluc (∼50,000 LU) and CXCR4-CFP (X4-CFP, ∼3,000–50,000 FU). As negative control, we used 5HT2B-Rluc (0.5 μg, ∼50,000 LU) (Lower). (B) BRET1 experiment scheme of the postulated interaction between CD4-Rluc and CCR5-YFP (Upper). We generated BRET titration curves by using 293T cells transiently cotransfected with CD4-Rluc as in A and CCR5-YFP (R5-YFP; ∼4,000–30,000 FU). We used 5HT2B-Rluc (0.5 μg, ∼50,000 LU) as negative control (Lower). BRET50 and BRETmax values (mean ± SEM) were calculated according to a nonlinear regression equation applied to a single binding-site model (n = 6) (ND, not determined).
BRET and bimolecular fluorescence complementation (BiFC) were combined to test whether CD4, CXCR4, and CCR5 organization is multimeric. The BiFC assay is a protein fragment-complementation method based on production of a fluorescent complex only when a protein–protein interaction is established (24). CXCR4/CCR5 heterodimerization was first tested by direct visualization of YFP in 293T cells transiently cotransfected with CCR5 fused to the N-terminal part of YFP (nYFP; amino acids 1–155) and CXCR4 fused to the C-terminal part of YFP (cYFP; 156–231) (Fig. 2 A and B). In a specificity control, fluorescence was negligible in 293T cells transiently cotransfected with CXCR4-cYFP and 5-HT2B-nYFP or with CCR5-nYFP and 5-HT2B-cYFP (Fig. 2 A and B). Correct CXCR4-cYFP, CCR5-nYFP, 5-HT2B-nYFP, and 5-HT2B-cYFP function was verified by measuring ligand-mediated Ca2+ flux for the chemokine receptors and agonist-triggered MAPK activation for the 5-HT2B constructs (Fig. S2 C and D). For BRET-BiFC assays, 293T cells were cotransfected with a constant amount of CD4-Rluc (donor) and increasing amounts of a 1:1 mixture of CXCR4-cYFP:CCR5-nYFP. The BRET signal was positive and increased as a hyperbolic function of the acceptor/donor ratio, confirming CD4/CXCR4/CCR5 oligomer formation (BRETmax 40.67 ± 4.90, BRET50 5.79 ± 1.81) (Fig. 2C). BRET was negligible when 5-HT2B-Rluc was used as donor.
Fig. 2.

CD4, CXCR4, and CCR5 form heterocomplexes. (A) We transiently cotransfected 293T cells with equal amounts of cDNA for CXCR4-cYFP (X4-cYFP) and CCR5-nYFP (R5-nYFP) fusion proteins, with CXCR4-cYFP and 5-HT2B-nYFP, or with 5-HT2B-cYFP and CCR5-nYFP fusion proteins. Fluorescence was determined at 530 nm; values represent mean ± SEM (n = 3, triplicates). Inset shows scheme of the postulated interaction between CXCR4-cYFP and CCR5-nYFP. (B) Confocal images at 48 h after transfection of the cells in A. (Scale bars: 20 μm). DIC, differential interference contrast microscopy. (C) Experiment scheme of CD4/CXCR4/CCR5 heterooligomer detection by BRET-BiFC (Upper). BRET-BiFC saturation curves (Lower) were obtained by using 293T cells cotransfected with 0.25 μg of cDNA for CD4-Rluc (∼75,000 LU) and increasing quantities of cDNA for X4-cYFP and R5-nYFP (0.2–3 μg, 14,000 FU). As negative control, cells were cotransfected with 0.5 μg of 5-HT2B-Rluc (∼100,000 LU). Curves were calculated according to a nonlinear regression equation applied to a single binding-site model. Values are mean ± SEM (n = 8). (D) Experiment scheme of CD4/CXCR4/CCR5 heterooligomer detection by SRET (Upper). We cotransfected 293T cells (Lower) with a constant amount of CD4-Rluc (∼50,000 LU) and X4-CFP (1 μg; 20,000 FU) and increasing R5-YFP quantities (0.2-1.5 μg, ∼60,000 FU). As a negative control, we cotransfected cells with 5-HT2B-Rluc (0.5 μg; ∼60,000 LU) and X4-CFP (1 μg; 20,000 FU), and increasing R5-YFP amounts (0.2–1.5 μg, 50,000 FU). Curves were calculated as in C. Values represent mean ± SEM (n = 8).
To confirm heterotrimerization, we used a sequential BRET/FRET technique (SRET) (25). We transiently cotransfected 293T cells with a constant amount of CD4-Rluc (BRET donor) and CXCR4-CFP (BRET acceptor and FRET donor), and increasing amounts of CCR5-YFP (FRET acceptor); the SRET signal was positive and saturable (SRETmax 197.1 ± 23.19, SRET50 18.53 ± 7.74) (Fig. 2D). Residual energy transfer was observed in control cells cotransfected with 5-HT2B-Rluc, CXCR4-CFP, and CCR5-YFP (Fig. 2D). These results indicate that CD4, CXCR4, and CCR5 form heterooligomers in living cells.
CCR5 Expression Alters CXCR4/CXCR4 Homodimeric and CD4/CXCR4 Heterodimeric Conformations.
To analyze the effect of CCR5 coexpression on CXCR4 homodimeric conformation, we transfected 293T cells with pcDNACCR5 or pcDNA, which we then cotransfected with constant amounts of CXCR4-CFP (donor) and increasing amounts of CXCR4-YFP (acceptor). CCR5 coexpression (Fig. S3A) did not significantly modify CXCR4-CFP or CXCR4-YFP fluorescence (Table 1). CCR5 expression significantly altered FRET50 for CXCR4 homodimers (CXCR4-CFP/CXCR4-YFP + pcDNA, 0.52 ± 0.02; CXCR4-CFP/CXCR4-YFP + pcDNACCR5, 1.13 ± 0.05; P < 0.05) (Fig. 3A), but not the total number of CXCR4 complexes (FRETmax). FRET50 reflects the apparent affinity of a given interaction (25, 26), and its variation can indicate conformational changes in the complex partners, which translate into longer or shorter donor-acceptor distances and/or changes in their relative orientation. Our results are nonetheless also compatible with CCR5-mediated interference with CXCR4 homodimers.
Table 1.
CXCR4-CFP (X4-CFP) and CXCR4-YFP (X4-YFP) fluorescence in 293T cells cotransfected with CCR5 or with empty vector (control) used for FRET saturation curves
| Plasmids | CXCR4-CFP (2.0 μg), FU | CXCR4-YFP, FU |
| X4-CFP/X4-YFP + control | 335,300 ± 152,600 | ∼100,000–1,600,000 |
| X4-CFP//X4YFP+CCR5 | 368,900 ± 122,000 | ∼300,000–2,300,000 |
Fluorescence was measured in a Wallac Envision 2104 reader for each sample before each FRET saturation curve experiment. Values represent mean ± SD fluorescence of donor expression (CXCR4-CFP) and increasing acceptor expression (CXCR4-YFP) in six experiments performed. FU, fluorescence units.
Fig. 3.
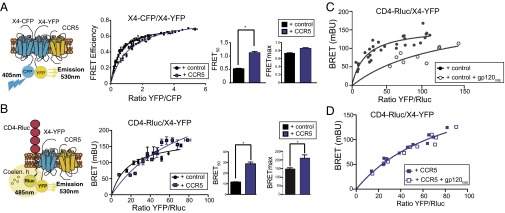
CCR5 alters CXCR4 homodimeric and CD4/CXCR4 heterodimeric conformations. (A) Experiment scheme used to evaluate by FRET the effect of CCR5 on CXCR4 homodimers (Left). FRET saturation curves (Center) by using 293T cells transiently cotransfected with CXCR4-CFP (X4-CFP) and CXCR4-YFP (X4-YFP) with pcDNA (control) or pcDNACCR5 (both 2 μg). FRET50 and FRETmax values were calculated by using a nonlinear regression equation for a single binding-site model and are expressed as mean ± SEM (n = 6) (Right). (B) Scheme of BRET1 experiment used to evaluate the effect of CCR5 on CD4/CXCR4 heterodimers (Left). We transiently transfected 293T cells with pcDNA or pcDNACCR5. Twenty-four hours after transfection, cells were cotransfected with a constant amount of CD4-Rluc and increasing amounts of X4-YFP (Center). BRET50 and BRETmax values were calculated as in A. Data are expressed as mean ± SEM (n = 5) (Right). (C and D) We transiently transfected 293T cells with pcDNA (control) (C) or pcDNACCR5 (CCR5) (D) as in B, and then stimulated with gp120IIIB (5 nM; 5 min, 37 °C). Data were analyzed by using a nonlinear regression equation as in B. One representative experiment of four is shown (*P < 0.05).
In subsequent BRET experiments, we tested whether CCR5 expression alters CD4/CXCR4 heterodimer conformation. Flow cytometry measurements showed similar membrane CCR5 expression in CCR5-expressing 293T cells cotransfected with constant amounts of CD4-Rluc (BRET donor) and increasing amounts of CXCR4-YFP (BRET acceptor) (Fig. 3B and Fig. S3B). Whereas CCR5 coexpression did not affect CD4-Rluc or CXCR4-YFP expression (Table 2), it significantly altered BRET saturation curves for CD4/CXCR4 heterodimers, as indicated by changes in BRETmax (CD4-Rluc/CXCR4-YFP + pcDNA, 148.6 ± 8.10; CD4-Rluc/CXCR4-YFP + pcDNACCR5, 211.6 ± 16.41) and in BRET50 (CD4-Rluc/CXCR4-YFP + pcDNA, 11.58 ± 2.43; CD4-Rluc/CXCR4-YFP + pcDNACCR5, 28.73 ± 6.76) (P < 0.05; Fig. 3B). These findings indicate that CD4/CXCR4 complexes form in the absence of ligand and that CCR5 incorporation into the heterooligomer alters CD4/CXCR4 complex conformation.
Table 2.
CD4-Rluc luminescence and CXCR4-YFP (X4-YFP) fluorescence in 293T cells cotransfected with CCR5 or with empty vector (control) used for BRET titration curves
| Plasmids | CD4-Rluc (0.65 μg), LU | CXCR4-YFP, FU |
| CD4-Rluc/X4-YFP + control | 13,238 ± 5,921 | ∼80,000–1,200,000 |
| CD4-Rluc/X4-YFP + CCR5 | 20,718 ± 5,033 | ∼250,000–1,400,000 |
Luminescence signal (CD4-Rluc) after coelenterazine H addition and fluorescence (CXCR4-YFP) were measured by using a Wallac Envision 2104 Reader. Values represent mean ± SD luminescence and fluorescence in five independent experiments.
CCR5 Alters gp120IIIB-Promoted CD4/CXCR4 Conformational Changes.
Conformational changes induced by gp120IIIB binding to CD4/CXCR4 were readily detected by BRET using CD4-Rluc as donor and CXCR4-YFP as acceptor in mock-transfected or CCR5-expressing 293T cells. CCR5, CD4, and CXCR4 levels were verified by FACS as above, and BRET curves were evaluated before and after incubation with soluble monomeric gp120IIIB (5 nM, 5 min, 37 °C). Paired analysis of the four groups of curves generated by CD4-Rluc/CXCR4-YFP + pcDNA and CD4-Rluc/CXCR4-YFP + pcDNACCR5, alone or with gp120IIIB (Fig. 3 C and D) using Akaike information criterion (n = 4) showed that the addition of gp120IIIB altered BRET saturation curves for CD4/CXCR4 heterodimers only when CCR5 was absent. These results show that gp120IIIB-triggered conformational changes in CD4/CXCR4 complexes are blocked by CCR5 coexpression.
CCR5 Blocks gp120IIIB-Mediated Early Actin Polymerization in CD4/CXCR4-Expressing Cells.
Shortly after binding to its receptors on resting CD4+ T cells, gp120 promotes rapid, transient polymerization of cortical actin (27, 28), a process that mimics the chemotactic response initiated by CXCL12 binding to CXCR4 (27–29). We tested the effect of gp120IIIB on actin in 293T cells expressing CD4/CXCR4 or CD4/CXCR4/CCR5. Phalloidin-FITC staining and flow cytometry data indicated that gp120IIIB triggered rapid actin polymerization (5–15 min) in CD4/CXCR4 but not in CD4/CXCR4/CCR5 cells (Fig. 4 A and B and Fig. S4A). Confocal microscopy analysis of this blockade in CD4/CXCR4 cells transiently transfected with CCR5-RFPm, incubated with gp120IIIB, and phalloidin-stained (Fig. S4 B and C) showed F-actin rearrangement in CD4/CXCR4 but not in CD4/CXCR4/CCR5 cells. As a control for cytoskeleton integrity, CXCL12 stimulation led to rapid polarized polymerization of cortical actin in both cell types (28, 29) (Fig. 4C and Fig. S4 D and E). Similar experiments were performed in primary CD4+ T cells nucleofected with CCR5 or the empty vector (Fig. 5A); flow cytometry showed that cell membrane CD4 and CXCR4 levels were unaffected by CCR5 expression (Fig. S5 A and B). As in CD4-expressing 293T cells, gp120IIIB promoted rapid actin polymerization (0.5–1 min) in CD4+ but not in CCR5+CD4+ T cells (Fig. 5B).
Fig. 4.

CCR5 blocks HIV gp120IIIB-mediated cortical actin dynamics in 293CD4 cells. (A and C) We transiently transfected 293CD4 cells with pcDNA (control; Left) or pcDNACCR5 (CCR5; Right) were treated with gp120IIIB (10 nM) (A) or CXCL12 (50 nM) (C), fixed, permeabilized, and stained with phalloidin-FITC for flow cytometry. A representative experiment is shown of five performed. (B) Quantitation of actin polymerization of cells in A. Data show mean ± SEM (*P < 0.05; n = 5).
Fig. 5.

gp120IIIB- and CXCL12-mediated actin dynamics in nucleofected CD4+ T cells. (A) Membrane expression of CCR5 in CD4+ T cells nucleofected with pcDNA (control) or pcDNACCR5 (CCR5) was determined by flow cytometry. (B and E) CD4+ T cells nucleofected with pcDNA or CCR5 were treated with gp120IIIB (10 nM) (B) or CXCL12 (50 nM) (E). At various times, treated cells were stained with anti-CCR5 and phalloidin-FITC. F-actin polymerization (%MFI phalloidin-FITC) was quantitated exclusively on CCR5+ cells. Data show mean ± SEM (n = 3). (C) F-actin (phallloidin-Alexa488, green) and CCR5 staining (anti-CCR5-Cy3, red) was visualized by confocal microscopy in CD4+ T cells nucleofected with pcDNA (control) or pcDNACCR5 (CCR5) and treated with gp120IIIB (10 nM, 1 min, 37 °C). Dashed line in DIC, differential interference contrast microscopy. Images indicates cell morphology (circular or elliptical). (Scale bars: 5 μm.) A representative image is shown (n = 3). Model used to calculate cell ellipticity with parameters (a, b, c), cell shape (dashed line), and the formula {eoblate = [2b2/(b2+c2)] x [1-2a2 − (b2+c2)]} are shown. (D) Quantitative analysis of confocal images for cell morphology (ellipticity) by actin cytoskeleton imaging in pcDNA- (control) and pcDNACCR5- (CCR5) nucleofected CD4+ T cells, alone (-) or treated (+) with gp120IIIB (C) (***P < 0.001). (F) Shape of CD4+ T cells nucleofected with pcDNA (control) or pcDNACCR5 (CCR5), treated with CXCL12 (50 nM, 1 min, 37 °C), and visualized by confocal microscopy. F-actin (phallloidin-Alexa488, green) and CCR5 (anti-CCR5-Cy3, red) staining. (Scale bars: 5 μm.) A representative image is shown (n = 3). (G) Quantitative analysis of CXCL12-promoted ellipticity using confocal images as in D (***P < 0.001).
Because HIV-1 gp120 binding modifies CD4+ T-cell shape (30, 31), we analyzed the gp120IIIB effect on morphology (ellipticity) by imaging the actin cytoskeleton in nucleofected CCR5+CD4+ and control CD4+ T cells. Fluorescence imaging of phalloidin-Alexa488 staining showed a rounded morphology for both cell types, with a relatively thin cortical actin layer (Fig. 5C and Fig. S6). Whereas incubation with gp120IIIB induced a change in control cell shape and formation of actin-rich protrusions, CCR5+CD4+ T cells were refractory to changes in shape (Fig. 5C and Fig. S6). In confocal images, quantitative analysis of the degree of deviation from a circular/spherical to an elliptical/ellipsoidal shape confirmed that these effects occurred only in primary CD4+ T cells (Imaris software; P < 0.001; Fig. 5D). Controls using CXCL12 showed cortical actin polymerization in both CD4+ and CCR5+CD4+ T cells (Fig. 5 E–G). These data show that in both the heterologous system and in primary CD4+ T cells, lack of gp120IIIB-triggered effects correlates with CCR5 expression.
CCR5 Expression in CD4/CXCR4 Cells Blocks gp120IIIB-Induced LIMK1 Activation and Cofilin Phosphorylation.
gp120-triggered actin polymerization involves transient LIMK1 activation, which phosphorylates and, thus, inactivates the actin depolymerization factor cofilin (27, 32). Cofilin phosphorylation by LIMK1 is also critical for CXCL12-induced actin reorganization and chemotactic responses in T lymphocytes (29). Whereas in CD4/CXCR4 cells gp120IIIB promoted rapid cofilin phosphorylation (Fig. 6A and Fig. S7A, Left), this effect was not detected in CD4/CXCR4/CCR5 cells (Fig. 6A and Fig. S7A, Right). CXCL12 nonetheless triggered cofilin phosphorylation in both cell types, which confirmed the integrity of the chemokine-mediated signaling machinery (Fig. 6A and Fig. S7B). In experiments using primary CD4+ T cells nucleofected with pcDNA or pcDNACCR5 (Fig. 6B), gp120IIIB induced rapid LIMK1 activation (30 s), followed by cofilin phosphorylation in CD4+ T cells (1 min) (Fig. 6C, Upper), but not in CD4+CCR5+ T cells (Fig. 6C, Lower). In both primary cell types, CXCL12 triggered LIMK1 and cofilin phosphorylation (Fig. 6D). These findings strongly suggest that CCR5 blocks gp120IIIB-triggered cytoskeletal reorganization events by altering the LIMK1/cofilin pathway.
Fig. 6.

CCR5 expression blocks gp120IIIB-mediated LIMK1 activation and cofilin phosphorylation. (A) We transiently transfected 293CD4 cells with pcDNA (control) or pcDNACCR5 (CCR5) were stimulated (1 min) with gp120IIIB (10 nM) or CXCL12 (50 nM). Densitometry data are shown as a mean ± SEM value of the corrected p-cofilin:GAPDH ratio (n = 5; *P < 0.05; **P < 0.01). (B) Flow cytometry analysis of CCR5 expression levels in CD4+ T cells nucleofected with pcDNA (control) or pcDNACCR5 (CCR5). (C and D) Cells in B were stimulated with gp120IIIB (10 nM) (C) or CXCL12 (50 nM) (D) at indicated times. Cell extracts were analyzed in Western blot with p-LIMK1 and p-cofilin mAb. Loading was controlled by reblotting for GAPDH. A representative experiment is shown (n = 3). Densitometry data are shown next to each image.
CCR5 Blocks HIV-gp120IIIB Binding to CD4/CXCR4.
To establish the mechanism involved in CD4/CXCR4/CCR5-mediated effects, we tested whether CCR5 coexpression altered gp120IIIB binding to CD4/CXCR4 complexes. A label-free surface plasmon resonance technology was used to study gp120IIIB biomolecular interactions with CD4, CXCR4, and CCR5 receptors expressed on lentiviral particles. Mock- and CCR5-transfected CD4+ 293 and 293T cells were transiently cotransfected with pLVTHM, PAX2, and VSVG plasmids to prepare lentiviral particles (LVP) bearing CD4/CXCR4, CD4/CXCR4/CCR5, CXCR4, or CXCR4/CCR5. We analyzed CXCR4 expression by flow cytometry, using latex beads that bind to LVP and specific antibodies (33); CD4 and CCR5 were evaluated by Western blot (Fig. S8 A and B). LVPCXCR4, LVPCXCR4/CCR5, LVPCD4/CXCR4, or LVPCD4/CXCR4/CCR5 were immobilized on the surface of a Biacore CM5 sensor chip, and gp120IIIB solutions (50–250 nM) were injected into each flow cell. We observed a dose-dependent response for gp120IIIB binding to LVPCD4/CXCR4, with maximum of ∼32 relative units (RU) for 250 nM and a minimum of ∼6 RU for 50 nM (Fig. 7A). In controls, binding to LVPCXCR4-coated and to LVPCXCR4/CCR5-coated flow cells was negligible. Sensorgrams were processed with Biaevaluation 4.1 software and adjusted to the 1:1 Langmuir binding model; kinetic parameters (kON = 4.7 ± 0.6 × 104 M⋅s−1 and kOFF = 2.0 ± 0.3 × 10−3⋅s−1) permitted calculation of the KD (44 nM) for gp120IIIB binding to LVPCD4/CXCR4 (Fig. 7G). We found no specific gp120IIIB binding to LVPCD4/CXCR4/CCR5 (Fig. 7 B and G).
Fig. 7.

CCR5 expression blocks gp120IIIB binding to CD4/CXCR4. (A–F) Sensorgrams for gp120IIIB binding (50–250 nM) to LVPCD4/CXCR4 (A) or LVPCD4/CXCR4/CCR5 (B) and for CXCL12 binding (50–250 nM) to LVPCXCR4 (C), LVPCXCR4/CCR5 (D), LVPCD4/CXCR4 (E), and LVPCD4/CXCR4/CCR5 (F) particles immobilized on a sensorchip. Aliquots of gp120IIIB or CXCL12 at distinct concentrations (50, 100, 150, 200, 250 nM) were injected sequentially into the flow cell, and binding was monitored as relative units (RU) on the sensorgram. The binding signal was subtracted for each gp120IIIB and CXCL12 concentration to the reference sensorchip. One representative sensorgram is shown of three obtained. (G) Association (KON) and dissociation (KOFF) constants (mean ± SEM, n = 3) of CXCL12 for LVPCXCR4, LVPCXCR4/CCR5, LVPCD4/CXCR4, LVPCD4/CXCR4/CCR5, and of gp120IIIB for LVPCD4/CXCR4 and LVPCD4/CXCR4/CCR5 were determined by fitting the data (A–F) using Biaevaluation 4.1 software (Biacore). KD = KOFF/KON. *Not available for the 1:1 Langmuir binding model.
To confirm this unanticipated observation, we carried out 125I-gp120IIIB binding assays to mock-transfected and CCR5-transiently transfected 293T (control) or CD4+ 293 cells. Scatchard analysis of gp120IIIB binding to CD4+ 293 cells, which express endogenous CXCR4, indicated a KD of 80 ± 8 nM, similar to previous reports (34). As predicted by our results above, specific gp120IIIB binding was not detected in CD4/CXCR4/CCR5 cells (Fig. S8D). As a control, CCR5 expression was verified by flow cytometry analysis (Fig. S8C). Control assays confirmed similar CXCL12 binding to CXCR4 in LVPCXCR4, LVPCXCR4/CCR5, LVPCD4/CXCR4, or LVPCD4/CXCR4/CCR5 (Fig. 7 C–G). The data indicate that CCR5 expression disrupted the gp120IIIB binding site in CD4/CXCR4 heterodimers, whereas it did not alter CXCL12 binding properties to CXCR4.
CCR5 Modulates CD4/CXCR4-Mediated Cell-Cell Fusion and X4 HIV-1 Infection.
The repeating unit in the HIV-1 envelope is a noncovalent trimer formed by gp120 and gp41 proteins (35). To test whether CCR5 expression also impairs trimeric gp120IIIB binding, we performed cell-cell fusion experiments by using 293T cells expressing gp120IIIB as effectors, and CD4+ 293 cells transiently expressing CCR5 (or mock-transfected) as target cells (Fig. 8A). Flow cytometry data confirmed that CD4+ 293 cells expressed endogenous CXCR4 and had physiological CD4 levels (36) (Fig. 8B). At 48 h after transfection, CCR5-expressing cells showed a ∼50% reduction in gp120IIIB-induced fusion compared with controls (Fig. 8C) (P < 0.01). To confirm that this effect is mediated by cell surface CCR5, we studied conditions for ligand (CCL5)-induced CCR5 internalization. CD4+ 293 cells stably transfected with CCR5 and treated (30 min) with CCL5 (100 nM) showed rapid CCR5 internalization (42 ± 3%), whereas expression of cell surface CD4 or CXCR4 was unaltered (Fig. S9A). CCL5-induced CCR5 internalization led to a significant increase in gp120IIIB-induced cell-cell fusion (Fig. 8C) (P < 0.05). These findings indicate that cell surface CCR5 reduces HIV-1 gp120IIIB-induced cell-cell fusion.
Fig. 8.

CCR5 coexpression reduces X4 HIV-1 entry in CD4/CXCR4 cells. (A) Flow cytometry analysis of CCR5 expression in target cells transfected (293CD4). As control, we used the same cells transfected with empty vector (pcDNA). (B) Endogenous expression as measured in flow cytometry of CXCR4 and CD4 after CCR5 transfection of target cells (293CD4). (C) Cell-cell fusion between 293TEnvIIIB effector and 293CD4 target cells transfected with CCR5. Target cells were stimulated with CCL5 (100 nM, 30 min) before cell-cell fusion. Data are expressed as the relative ratio of Env-induced fusion, using 293T cells as reference. Data show mean ± SEM of five experiments in triplicate (**P < 0.01; *P < 0.05; two-tailed Mann–Whitney nonparametric t test).
To test the effect of CCR5 expression in viral particles bearing native gp120/gp41 complexes, Jurkat CD4 cells or primary CD4+ T cells were nucleofected by using pcDNA or pcDNACCR5 plasmids (Fig. 9 A and B and Fig. S5) and incubated with X4 HIV-1NL4-3 strain virus. At 48 h after infection, ELISA measurement of p24 in culture medium showed that CCR5 expression reduced HIV-1 infection in both cell models (Fig. 9 C and D) (P < 0.05 in Jurkat cells; P < 0.01 in primary T cells). These results indicate that CCR5 regulates X4 HIV-1 entry into CD4+ T cells.
Fig. 9.

CCR5 effect on HIV-1 infection in Jurkat and CD4+ T cells. (A and B) Flow cytometry analysis of membrane levels of CCR5 in Jurkat (A) or CD4+ T cells (B) nucleofected with pcDNA (control) or pcDNACCR5 (CCR5). (C) Jurkat CD4 cells as in A, untreated or treated with CXCL12 (100 nM, 15 min) as indicated, were incubated with the X4 HIV-1NL4-3 (Left) or the R5 HIV-1NLAD8 strain (Right) and viral infection determined. CCR5 expression significantly reduced X4 HIV-1 infection of Jurkat cells (*P < 0.05) and CXCR4 expression significantly affected R5 HIV-1 infection (*P < 0.05). Data show mean ± SEM from four independent experiments in quadruplicate. (D) Viral infection of CD4+ T cells nucleofected with CCR5 was significantly reduced compared with control (CD4+ T cells nucleofected with pcDNA) (**P < 0.01). Data show mean ± SEM from five independent experiments in quadruplicate.
We determined whether the CD4/CXCR4/CCR5 complexes also affect R5 virus infection. Jurkat cells transfected with pcDNACCR5 plasmid as above (Fig. 9A) were infected by the R5 HIV-1 strain NLAD8 (Fig. 9C). Because CXCR4 is expressed constitutively in these cells, we reduced CXCR4 levels by CXCL12-triggered internalization before testing R5 HIV-1NLAD8 infection. In cells in which surface CXCR4 levels were reduced by CXCL12 (∼80%, 15 min) without altering CCR5 or CD4 levels (Fig. S9B), R5-HIV-1NLAD8 infection was significantly higher than in untreated cells (Fig. 9C; P < 0.05). These data confirm the influence of the CXCR4/CCR5 ratio for HIV-1 infection.
Discussion
For more than a decade, chemokine receptors have been known to preexist on cells as homooligomers and heterooligomers (18, 19, 26, 35, 37, 38). Although heterodimer stabilization is associated with specific signaling events (39–41) and with modulation of individual receptor activity (36, 42, 43), the functional relevance of these complexes remains unclear. This fact is the case of the two main HIV-1 coreceptors, CXCR4 and CCR5. When coexpressed on a cell and in the absence of ligands, these two receptors form heterodimers (39, 40, 44) that appear to modulate lymphocyte functions (40). This effect is compatible with the consensus for the G protein-coupled receptors (GPCR), which considers heteromers as entities whose function differs from that of the individual receptors (45).
Although GPCR oligomerization is reported, there are few examples of complexes that include more than two receptor proteins; one is that of the cannabinoid CB1/dopamine D2/adenosine A2A receptor oligomers identified by SRET (25). Using two energy transfer approaches, BRET-BiFC and SRET, we identified heterocomplexes formed by two members of the GPCR family (CXCR4 and CCR5) and one of the Ig superfamily (CD4). In addition, CCR5 coexpression promoted significant FRET50 variation in CXCR4 homodimers without altering FRETmax values; this finding indicated that CCR5 did not affect the number of CXCR4 complexes, but modulated the apparent affinity between the two CXCR4 partners (46, 47), although we cannot rule out CCR5 interference with CXCR4 homodimer formation. Such modifications reflect CCR5-mediated alterations in CXCR4 complexes. CCR5 expression also reduced FRET50 and increased FRETmax of CD4/CXCR4 heterodimers, that is, it affected both the apparent affinity between CD4 and CXCR4 and the number of complexes on the cell (48) because of changes in the distance and/or the orientation of the partners. This effect, and the ability of CD4, CXCR4, and CCR5 to form trimeric complexes, rules out competition by the two chemokine receptors for CD4 association.
Conformational rearrangement of the partners in a heterodimeric complex can alter ligand binding (49, 50) and affect ligand function (42, 51) by modulating their ability to activate G proteins (43, 50). Here, we show evidence that CCR5 coexpression and receptor oligomerization impede gp120IIIB binding to target cells. The CCR5 effect on gp120IIIB binding and function is specific, as CXCL12-mediated signaling events were unaffected. It is thus possible that the CD4 domains involved in gp120IIIB binding are masked by the CD4/CXCR4/CCR5 oligomer. When gp120 binding to CD4 is prevented, the gp120IIIB conformational changes necessary for subsequent CXCR4 binding (52) did not take place. Our BRET data (Fig. 3 C and D) showed a gp120IIIB-promoted conformational change in CD4/CXCR4 that was blocked by CCR5 coexpression, reinforcing the idea that gp120IIIB is unable to bind to CD4/CXCR4/CCR5 complexes.
CCR5 coexpression also impaired the cell-cell fusion that allows HIV-1 entry into target cells, indicating that the effect observed using soluble monomeric gp120IIIB is also evident in virus-bearing native trimeric gp120 and gp41 protein complexes. One of the earliest events in HIV-1NL4-3 infection is CXCR4-mediated activation of LIMK1 and cofilin phosphorylation; these events increase cortical actin dynamics in resting CD4+ T cells (27, 32), a process that might require CXCR4 dimerization (53). Our data confirm these observations, because we detected rapid gp120IIIB-mediated actin polymerization following transient LIMK1 activation and cofilin inactivation. This chain of events, necessary to trigger membrane fusion and viral entry, was blocked when cells coexpressed CCR5. Although gp120IIIB-mediated actin polymerization, LIMK1 activation, and cofilin phosphorylation were abolished, inhibition of cell-cell fusion and HIV-1NL4-3 infection was incomplete. This discrepancy is probably due to differences in the cell types analyzed. We determined viral infection by using the entire population of CCR5-transfected CD4+ primary T or Jurkat CD4+ cells (40% transfection efficiency), whose expression of CCR5 resembled that of activated primary T cells and Th1 cells (54, 55), whereas our analysis of the mechanism involving actin polymerization was restricted to CCR5+ cells. Our data concur with a report that, in NIH 3T3 cells coexpressing CD4, CXCR4, and CCR5, the T-tropic HIV-1 isolate HCF was less infective than in CCR5-negative cells (56); another study showed lower infectivity of primary X4 viruses (ELI 1 and K4) in HeLa-CD4 cells when CCR5 was coexpressed (57). HIV-1NL4-3 replication is also higher in peripheral blood mononuclear cells from CCR5-Δ32 heterozygous donors than from controls (58). Our study shows that CCR5 coreceptor expression reduced X4 HIV-1 entry into cells and infection, and describes the molecular mechanism involved (Fig. 10 A and B).
Fig. 10.

Effect of CCR5 on X4 HIV-1 infection. (A) CD4+ T cells express CXCR4 and CD4, which form homodimers and heterodimers at the cell surface. X4 gp120 binding to CD4 creates binding sites on CXCR4 and activates LIMK-1, which, in turn, phosphorylates cofilin and facilitates HIV-1 infection. (B) CCR5-coexpressing cells form CD4/CXCR4/CCR5 oligomers, which modifies CD4/CXCR4 and CXCR4/CXCR4 conformation and impedes X4 gp120 binding and HIV-1 infection. (Inset) CD4 C1 is the CD4 conformation in the absence of CCR5; CXCR4 C1 is the CXCR4 conformation in the absence of CCR5; CD4 C2 is the CD4 conformation in the presence of CCR5; and CXCR4 C2 is the CXCR4 conformation in the presence of CCR5.
There is negative selection for X4 viruses in patients, and the R5 are the most commonly transmitted HIV-1 strains (59). Macrophages, dendritic and CD4+ T cells that express CCR5 and, to a lesser extent CXCR4, are the main immune cells in genital and rectal subepithelial tissue and in gut-associated lymphoid tissue (60–62). The situation differs in blood, where CCR5 expression is restricted to 5% of circulating immune cells, most of them memory T cells, i.e., 15% of CD4+ T cells (63). CCR5-expressing cells thus concentrate at primary infection sites, which could explain why R5 virus entry prevails over those that use CXCR4. Nonetheless, although X4 viruses do not enter, immune cells at the mucosa express CXCR4. CD4/CXCR4/CCR5 oligomerization could be a dynamic way to explain these apparent discrepancies. We observed that CCR5 levels at the CD4+CXCR4+ cell membrane determined X4 HIV-1 infection. Furthermore, reduction of CXCR4 expression in these cells increased R5 HIV-1 infection. R5 viruses are associated with the asymptomatic phase of AIDS, which coincides with acute infection and affects mainly Th1 cells that express surface CCR5. The switch from R5 to X4 viruses is associated with the loss of CD4+ T cells and correlates with Th2 cell infection (13, 14) and AIDS development (symptomatic phase). Th1 and Th2 cells have similar CXCR4 levels, whereas Th2 cells express little CCR5 (55, 64). During this symptomatic phase, early CXCR4 and CD4 expression during T-cell development in the thymus renders these cells susceptible to X4 HIV-1 infection and, thus, promotes a defect in immune system regenerative capacity that exacerbates AIDS (65). These data suggest that the HIV-1 coreceptor ratio influences cell susceptibility to infection and contributes to viral pathogenesis.
Our results also indicate that receptor heterooligomerization increases cell plasticity, which must be considered when evaluating the functional and pharmacological effects of drugs that act on GPCR and when exploring new therapeutic approaches for blocking HIV-1 binding and infection. Compounds engineered to mimic the CCR5-triggered conformational changes in CXCR4 homodimers or CD4/CXCR4 heterodimers could reduce virus-induced damage to the immune system, making them suitable for blocking X4 HIV-1 infection.
Methods
Cells and Reagents.
HEK293T (293T) cells were obtained from the American Type Culture Collection (CRL-11268). HEK293CD4 (293CD4) cells were generated in the M.M. laboratory, and CNB/CSIC (36) and the stable cell line HEK293CD4/CCR5 was derived from 293CD4 cells. Jurkat CD4 cells were donated by J. Alcamí (Centro Nacional de Microbiología, Inst Salud Carlos III, Madrid, Spain). Human lymphocytes were isolated from healthy donor blood by centrifugation through Percoll density gradients [1,800 × g, 45 min, room temperature (RT)], and CD4+ cells were purified by negative selection using Dynabeads (Invitrogen Dynal).
We used anti-human CCR5 (CTC8, R&D); anti-human CD4 (OKT-4; eBioscience) and biotin-anti-human CXCR4B (12G5, R&D); biotin-anti-human CD4-FITC (RPA-T4, BD) and biotin-anti-CCR5-PE (2D7, BD) or biotin-anti-CCR5-FITC (2D7, BD); Cy3-goat anti-mouse IgG, streptavidin-SPRD (Jackson ImmunoResearch); phalloidin-FITC and phalloidin-Alexa488 (Sigma-Aldrich); anti-phospo-LIMK1(Thr508)/LIMK2(Thr505); anti-phospho-cofilin (pS3) (77G2; Cell Signaling); and GAPDH (Santa Cruz Biotechnology). CXCL12 and CCL5 were from PeproTech and recombinant HIV-1 IIIB glycoprotein gp120 (CHO) from ImmunoDiagnostics. Jet PEI (Polyplus Transfection) was used to transiently transfect 293T cells, except for FRET saturation curves. Plasmids (10 μg) were nucleofected into Jurkat cells with a BioRad electroporator [20 × 106 cells/400 μL of RPMI 1640 with 10% (vol/vol) FCS]. CD4+ T cells were nucleofected by using Amaxa kits for human T cells (Amaxa) and used 24 h after transfection. Positive nucleofected cells ranged from 40 to 90%.
Fusion Proteins and Expression Vectors.
The N-terminal truncated YFP (nYFP; amino acids 1–155) and the C-terminal truncated YFP (cYFP; amino acids 156–231) vectors, as well as pEYFP-N1-mGluR1a and pRluc-N1-5-HT2B plasmids, were generated in the R.F. laboratory, Universidad Autónoma de Barcelona. Human CXCR4 and CCR5 receptors were PCR amplified from pcDNA3.1-CXCR4 and pcDNA3.1-CCR5 (pcDNACCR5) by using oligonucleotides listed below and cloned into pECFP-N1, pEYFP-N1, pERFPm-N1 (Clontech Laboratories), cYFP, and nYFP. pECFP-N1/pEYFP-N1 for X4: 5′HindIII (5′ATAAGCTTAT GGAGGGGATCAGTATATACATTC3′) and 3′AgeI (5′GACCGGTGGATCCCGTAAGCT GGAGTGAAAACTTGAAG3′); cYFP/nYFP 5′NheI (5′GCTAGCATGGAGGGGATCAGT ATATACAC3′) and 3′EcoRI (5′GAATTCTAAGCTGGAGTGAAAACTTGAAG3′). pECFP-N1/pEYFP-N1/pERFPm-N1 for R5: 5′HindIII (5′TAAAGCTTATGGATTATCAAG TGTCAAGTCC3′) and 3′AgeI (5′GACCGGTAATAACAAGCCCACAGATATTTC3′) and for cYFP/nYFP 5′NheI (5′AAGCTAGCATGGATTATCAAGTGTCAAGTCC3′) and 3′EcoRI (5′GAATTCTAACAAGCCCACAGATATTTCC3′).
Human CD4 was cloned by PCR from T lymphocytes by using the oligonucleotides listed below and cloned into pRluc-N1 (Perkin-Elmer): 5′XhoI (5′TTCTCGAGATGAACCGGGG AGTCCCTTTTAG3′) and 3′HindIII (5′AAGCTTTAAAATGGGGCTACATGTCTTCTG3′).
Human 5-HT2B was PCR-amplified from 5-HT2B-YFP by using the following oligonucleotides, then cloned into pcDNA3-cYFP and pcDNA3-nYFP: 5′NheI (5′TTTGCTA GCATGGCTCTCTCTTACAGAGTGTC3′) and 3′KpnI (5′GGTACCATACATAACTAAC TTGCTCTTAG3′).
FRET Analysis.
We cotransfected 293T cells and obtained FRET saturation curves as described (26). To establish the influence of CCR5 expression on CXCR4/CXCR4 homodimers or CD4/CXCR4 heterodimers in FRET saturation curves, we first transiently transfected 293T cells with cDNA encoding the fusion proteins with pcDNA3.1 (pcDNA) or pcDNACCR5 (24 h). To determine FRET50 and FRETmax values, curves were extrapolated from data by using a nonlinear regression equation applied to a single binding site model with a 95% confidence interval (GraphPad PRISM 5.0).
BRET.
We transiently cotransfected 293T cells with a constant amount (0.65 μg) of cDNA encoding CD4-Rluc [50,000–100,000 luminescence units (LU)] and increasing amounts of cDNA for X4-CFP (0.2–1.5 μg) for BRET2, or X4-YFP (0.2–1.5 μg) or R5-YFP (0.2–1.8 μg) for BRET1. Fluorescent proteins (20 μg) were quantified by using the Wallac Envision 2104 Multilabel Reader (PerkinElmer) equipped with a high-energy xenon flash lamp (X4-CFP, 8-nm bandwidth excitation filter at 405 nm; X4-YFP and R5-YFP, 10 nm bandwidth excitation filter at 510 nm). Receptor fluorescence expression was determined as fluorescence of the sample minus the fluorescence of cells expressing CD4-Rluc alone. For BRET2 and BRET1 measurements, the equivalent of 20 μg of cell suspension was distributed in 96-well microplates (Corning 3912; flat-bottom white plates), followed by 5 μM DeepBlueC (BRET2) (Biotium) or coelenterazine H (BRET1) (PJK). For BRET2 experiments, signals were obtained immediately after DeepBlueC addition (30 s) by using the Wallac Envision 2104 Reader, which allows integration of signals detected in the short-wavelength filter (8 nm bandwidth, 405 nm) and the long-wavelength filter (10 nm bandwidth, 486 nm). For BRET1, readings were collected 1 min after coelenterazine H addition, because the Wallac Reader allows integration of signals detected in the short- (10 nm bandwidth, 510 nm) and long-wavelength filters (10 nm bandwidth, 530 nm). Receptor-Rluc luminescence signals were acquired 10 min after coelenterazine H (5 mM) addition. BRET is defined as [(long wavelength emission)/(short wavelength emission)]−Cf, where Cf is [(long wavelength emission)/(short wavelength emission)] for the Rluc construct expressed alone in the same experiment.
To determine the influence of CCR5 expression on CD4/CXCR4 heterodimers, we transfected 293T cells with pcDNA (control) or pcDNACCR5 (CCR5); after 24 h, we transiently transfected the cells with cDNA encoding CD4-Rluc/CXCR4-YFP. In similar experiments, we evaluated the effect of gp120IIIB stimulation (5 nM, 5 min, 37 °C). Curves in these groups (CD4-Rluc/CXCR4-YFP + pcDNA and CD4-Rluc/CXCR4-YFP + pcDNACCR5) were paired, and dimerization in the same group of cells was evaluated before and after gp120IIIB addition. To determine which model best fit the data for the four pairs of saturation curves (n = 4), we used Akaike information criterion corrected for small sample size (AICc) [simpler model: one curve for all datasets (before and after gp120IIIB stimulation); alternative model: different curves for each dataset] (66). If the majority of the AICc difference (Δ) is positive, the preferred model is a distinct curve for all datasets; if Δ is negative, the preferred model is one curve for all datasets. We used GraphPad PRISM 5.0 software.
For BRET assays using BiFC, we cotransfected 293T cells with a constant amount of cDNA encoding CD4-Rluc or 5-HT2B-Rluc receptor and increasing amounts of a cDNA mixture encoding CXCR4-cYFP and CCR5-nYFP (1:1, cYFP:nYFP); fluorescence complementation and BRET were determined as above. Fluorescence and luminescence were measured for each sample before each experiment to confirm similar donor expression (∼75,000 LU) while monitoring the increase in acceptor expression [2,000–14,000 fluorescent units (FU) for complemented YFP]. In each BRET saturation curve, the relative amount of acceptor is given by the ratio between acceptor fluorescence (YFP) and donor luciferase activity (Rluc).
SRET.
We transiently cotransfected 293T cells with distinct amounts of plasmids encoding fusion proteins (CD4-Rluc or 5-HT2B-Rluc, CXCR4-CFP, and CCR5-YFP). Using aliquots of transfected cells (20 μg of protein), we performed three experiments in parallel. For the first, protein-YFP expression was determined by detection of protein-YFP fluorescence. Cells were distributed in 96-well microplates (transparent-bottom black plates) and read in a Fluostar Optima Fluorimeter equipped with a high-energy xenon flash lamp, using an excitation filter at 485 nm and 10-nm bandwidth emission filters corresponding to 510 nm (506–515 nm; channel 1) and 530 nm (527–536 nm; channel 2). As for FRET, we separated the relative contribution of the fluorophores to the detection channels for linear unmixing. We measured the contribution of CFP and YFP proteins alone to the two detection channels (spectral signature) in cells expressing only one of these proteins and normalized to the sum of the signal obtained in the two detection channels. Fluorescence was calculated as the difference between the fluorescence of cells expressing only protein-Rluc and those expressing protein-YFP.
In the second experiment, protein-Rluc expression was quantified by detecting its luminescence. Cells were distributed in 96-well microplates (Corning; flat-bottom white plates), and the luminescence signal was determined 10 min after coelenterazine H (5 μM) addition, in a Mithras LB 940 multimode reader (Berthold Technologies). Finally, for SRET measurements, cells in 96-well microplates (Corning; white-bottom white plates) were incubated with DeepBlueC (5 μM) and the SRET2 signal detected in a Mithras LB 940 reader with detection filters for short [400 nm (370–430 nm)] and long wavelength [530 nm (510–560 nm)]. By analogy with BRET, we defined net SRET as [(long-wavelength emission)/(short-wavelength emission)] − Cf, where Cf is [(long-wavelength emission)/(short-wavelength emission)] for cells expressing protein-Rluc, protein-CFP, or protein-YFP. Linear unmixing was done for SRET2 quantification only, considering the spectral signature as described above to separate the two fluorescence emission spectra.
Cell-Cell Fusion Assay.
Stable 293CD4 cells (which express endogenous CXCR4) were cotransfected with the pSCluc plasmid bearing the firefly luciferase gene under the control of the vaccinia virus 7.5 promoter, the promoterless renilla luciferase plasmid (pRNull) and, when needed, with increasing amounts of cDNA encoding CCR5 (target cells). HIV-1envIIIB was introduced into effector 293T cells by infection with recombinant vaccinia virus (1 h, 37 °C). At 12 h after infection, 105 effector cells cultured in 100 μg/mL rifampicin were mixed with target cells (6 h, 37 °C), and cell-cell fusion was analyzed by measuring luciferase/renilla activity in lysates by using the Dual-Glo Luciferase Assay System (Promega). We cotransfected 293T target cells with pcDNACCR5 or empty vector (pcDNA3.1) and pSCluc; pRNull plasmids were used as controls. Luciferase activity was calculated as the relative ratio (firefly luminescence activity/renilla luminescence)/(control firefly luminescence activity/control renilla luminescence).
Flow Cytometry Analysis.
Cells were plated in V-bottom 96-well plates (2.5 × 105 cells per well) and incubated with specific antibodies (30 min, 4 °C), followed by flow cytometry. Cell-bound fluorescence was determined in a Profile XL or Gallios flow cytometer (Beckman Coulter). Chemokine receptor expression was quantified by using a Dako Qifikit (DakoCytomation) (67). For internalization analysis, 293CD4/CCR5 cells (2 × 105 per well) were stimulated with CCL5 for various times (100 nM, 37 °C) and the reaction terminated with cold PBS before cytometry analysis as above.
Phalloidin-FITC Staining of F-Actin and Flow Cytometry.
We stimulated 293CD4 (106) or nucleofected CD4+ T (2 × 106) cells with gp120IIIB (10 nM) or CXCL12 (50 nM) at 37 °C. Cells were pelleted, paraformaldehyde-fixed (4% PFA; 10 min, RT), washed three times with cold PBS, and stained with violet LIVE/DEAD Fixable Dead Cell Stain (Molecular Probes) to identify live CD4+ T cells. When needed, cells were stained with anti-CCR5-PE (30 min, 4 °C). All cells were stained with phalloidin-FITC in permeabilization buffer (0.1% Triton ×100, 1% BSA, 0.1% goat serum, and 50 mM NaCl in PBS; 30 min, 4 °C). Stained cells were analyzed on a Gallios flow cytometer. For CD4+ T cells nucleofected with pcDNACCR5, F-actin polymerization was determined exclusively for CCR5+ cells.
Immunofluorescence.
Nucleofected primary CD4+ T cells (2 × 105 per well) were cultured on fibronectin-coated (20 μg/mL) Teflon-printed slides (Electron Microscopy Sciences; 30 min, 37 °C), then treated with gp120IIIB (10 nM) or CXCL12 (50 nM). Cells were washed in cold PBS and fixed with 4% PFA (10 min, RT). To prevent nonspecific binding, cells were treated with PBS with 1% BSA, 0.1% goat serum, and 50 mM NaCl (30 min, 37 °C). CD4+ T cells nucleofected with pcDNACCR5 were stained with anti-CCR5 mAb (30 min, RT), followed by Cy3-goat anti-mouse IgG (20 min, RT). After washing, cells were incubated with phalloidin-Alexa488 in permeabilization buffer (30 min, RT), slides were mounted with Fluoromount-G medium (Southern Biotech), and fluorescence evaluated on an Olympus IX81 microscope with a PLAPON 60 × 03 objective (aperture 1:40) and FV10-ASW 1.6 software. Samples were excited with two laser lines (Alexa488, 488 nm: Cy3, 543). The SDM560 dichroic mirror was used for double staining; filters used were 500–530 nm for Alexa488 and 555–655 nm for Cy3. All images were processed with ImageJ and ellipticity analyzed with Imaris 7.0 software (Bitplane AG).
Western Blot.
Cells were lysed in detergent buffer (1% Nonidet-P40, 50 mM Tris⋅HCl at pH 8.0, 150 mM NaCl, 0.5 mM EDTA, 10 mM sodium pyrophosphate, 1 mM PMSF, 10 μg/mL aprotinin, 10 μg/mL leupeptin, and 10 mM sodium orthovanadate; 30 min, 4 °C). Protein extracts (20–50 μg) were separated by 10–12% SDS/PAGE and transferred to a nitrocellulose membrane. After blocking, membranes were incubated with primary antibodies (anti-CCR5, anti-CD4, anti-p-cofilin, anti-p-LIMK1 and anti-GAPDH; 4 °C, overnight), followed by horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit antibodies (Southern Biotech; 45 min, RT), and developed with the enhanced ECL detection system. Blots were quantified by using ImageJ.
Virion Production, Purification, and Characterization.
Lentiviral particles were produced by JetPei cotransfection of 293T or 293CD4 cells with LVTHM/GFP, PAX2, and VSVG plasmids (Tronolab) at a 1:1:1 ratio. When necessary, pcDNACCR5 was cotransfected by using JetPei 24 h before transfection with viral plasmids. At 72 h after transfection, supernatant was harvested and cell debris removed by low-speed centrifugation and 0.45-μm filtration. The supernatant was pelleted in a Beckman SW55 rotor (247,000 × g, 2 h, 4 °C) through a 20% sucrose cushion and the pellet was resuspended in PBS. Several batches of lentiviral particles were standardized by titration using 293T cell transduction with twofold serial dilutions of viral particles; after 72 h, GFP expression was analyzed by FACS. Lentiviral particles with a similar titration index were aliquoted and stored at −80 °C.
Immobilization of Lentiviral Particles on a Sensor Chip and Biacore Kinetic Assays.
Equal volumes of 0.1 M N-Hydroxysuccinimide and 0.4 M 1-ethyl-3-3-dimethylaminopropyl carbodiimide hydrochloride were mixed and injected (5 μL/min, 7 min, RT) over the surface of a CM5 sensor chip (GE Healthcare) to activate the carboxymethylated dextran. Hepes-buffered saline (HBS-P) [10 mM Hepes, 0.15 M NaCl, and 0.005% polyoxyethylenesorbitan (P20) at pH 7.4] was used as immobilization running buffer. Lentiviral particles (107/mL) diluted in sodium acetate buffer (10 mM, pH 4.0) were injected over the activated surfaces (5 μL/min, 7 min, RT), followed by ethanolamine (1 M, pH 8.5, 5 μL/min, 7 min, RT) to deactivate remaining active carboxyl groups. All determinations were made by using a Biacore 3000 (GE Healthcare). gp120IIIB (50–250 nM) or CXCL12 (50-250 nM) in PBS-P buffer (137 mM NaCl, 10 mM Na2HPO4, 1.76 mM KH2PO4, 2.7 mM KCl, 0.005% P20, pH 7.4) was injected over immobilized viral particles (30 μL/min, 2 min, 25 °C; association phase), followed by a 4-min injection of Tris buffer alone over the surface (dissociation phase). Sensorgrams were corrected for background signals in the reference flow channels (a chamber with immobilized LVPCXCR4 or LVPCXCR4/CCR5 particles after gp120IIIB injection, and an empty chamber activated and deactivated in parallel by CXCL12 injection). Kinetic assays were followed by injection of 5 mM HCl to dissociate remaining ligand (regeneration). All steps were performed by using the system’s automated robotics; all phases were followed in real time as a change in signal expressed in relative units. Curves derived from these assays were used to generate kinetic constants, and analyzed by fitting to a simple one-site interaction model with Biaevaluation 4.1 software (Biacore). Alternatively, dissociation constants were derived from the response at equilibrium to corroborate findings from automated kinetic analyses. KON, KOFF, and KD were analyzed by using ANOVA, followed by the nonparametric Kruskall–Wallis test for multiple comparisons (GraphPad, PRISM 5.0). For more information, see SI Methods.
Virus Preparation and Infection.
The X4 HIV virus strain NL4-3 was obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH): pNL4-3 from M. Martin (Laboratory of Molecular Microbiology, NIAID, NIH, Bethesda) (48) and the R5 HIV-1 virus strain NLAD8 from E. Vacas (Hospital General Universitario Gregorio Marañón, Madrid, Spain). For p24 production, nucleofected CD4+ T cells and Jurkat CD4 cells were infected with 20 ng of HIV-1NL4-3/106 cells or with 50 ng of HIV-1NLAD8/106 cells (2 h, 37 °C; equivalent to 1–2 multiplicity of infection or viral particles per cell), then washed extensively with medium to remove free viral particles. Infected and uninfected cells were maintained in culture (24 h, 37 °C), supernatants were harvested, and p24 concentration was measured by ELISA (Innotest HIV-1 antigen mAb; Innogenetics).
Statistical Analyses.
Results were analyzed by using GraphPad PRISM 5.0 (***P < 0.001, **P ≤0.01, *P <0.05). We used an unpaired two-tailed Student’s t test to compare two subject groups and the two-tailed Mann–Whitney test for correlation analysis of FRET by acceptor photobleaching. Data are given as mean ± SEM.
Supplementary Material
Acknowledgments
We thank S. Álvarez and L. Díaz for technical support, C. Bastos for secretarial assistance, and C. Mark for editorial assistance. This work was supported in part by Spanish Ministry of Science and Innovation Grant SAF 2011-27370, RETICS (Redes Temáticas de Investigación Cooperativa en Salud) Program Grants RD08/0075/0010 and RD12/0009/009 (RIER, Red de Inflamación y Enfermedades Reumáticas), Madrid regional government Grant S2010/BMD-2350 [Rheumatoid Arthritis: Physiopathology mechanisms (RAPHYME)], and European Union 7th Framework Programme for Research and Technological Development (FP7)-integrated project Masterswitch 223404.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322887111/-/DCSupplemental.
References
- 1.Klatzmann D, et al. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984;312(5996):767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 2.Alkhatib G, et al. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272(5270):1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 3.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272(5263):872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 4.Basmaciogullari S, Pacheco B, Bour S, Sodroski J. Specific interaction of CXCR4 with CD4 and CD8alpha: Functional analysis of the CD4/CXCR4 interaction in the context of HIV-1 envelope glycoprotein-mediated membrane fusion. Virology. 2006;353(1):52–67. doi: 10.1016/j.virol.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 5.Lapham CK, Zaitseva MB, Lee S, Romanstseva T, Golding H. Fusion of monocytes and macrophages with HIV-1 correlates with biochemical properties of CXCR4 and CCR5. Nat Med. 1999;5(3):303–308. doi: 10.1038/6523. [DOI] [PubMed] [Google Scholar]
- 6.Lee S, et al. Coreceptor competition for association with CD4 may change the susceptibility of human cells to infection with T-tropic and macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 2000;74(11):5016–5023. doi: 10.1128/jvi.74.11.5016-5023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaitseva M, et al. Increased CXCR4-dependent HIV-1 fusion in activated T cells: Role of CD4/CXCR4 association. J Leukoc Biol. 2005;78(6):1306–1317. doi: 10.1189/jlb.0105043. [DOI] [PubMed] [Google Scholar]
- 8.Xiao X, et al. Constitutive cell surface association between CD4 and CCR5. Proc Natl Acad Sci USA. 1999;96(13):7496–7501. doi: 10.1073/pnas.96.13.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loetscher P, et al. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;391(6665):344–345. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- 10.Bonecchi R, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187(1):129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haynes BF, Pantaleo G, Fauci AS. Toward an understanding of the correlates of protective immunity to HIV infection. Science. 1996;271(5247):324–328. doi: 10.1126/science.271.5247.324. [DOI] [PubMed] [Google Scholar]
- 12.Salk J, Bretscher PA, Salk PL, Clerici M, Shearer GM. Response. Science. 1993;262(5136):1075–1076. doi: 10.1126/science.262.5136.1075. [DOI] [PubMed] [Google Scholar]
- 13.Tersmette M, et al. Differential syncytium-inducing capacity of human immunodeficiency virus isolates: Frequent detection of syncytium-inducing isolates in patients with acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. J Virol. 1988;62(6):2026–2032. doi: 10.1128/jvi.62.6.2026-2032.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu T, et al. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science. 1993;261(5125):1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]
- 15.Clerici M, Shearer GM. A TH1—>TH2 switch is a critical step in the etiology of HIV infection. Immunol Today. 1993;14(3):107–111. doi: 10.1016/0167-5699(93)90208-3. [DOI] [PubMed] [Google Scholar]
- 16.Romagnani S. Human TH1 and TH2 subsets: Regulation of differentiation and role in protection and immunopathology. Int Arch Allergy Immunol. 1992;98(4):279–285. doi: 10.1159/000236199. [DOI] [PubMed] [Google Scholar]
- 17.Naif HM, et al. CCR5 expression correlates with susceptibility of maturing monocytes to human immunodeficiency virus type 1 infection. J Virol. 1998;72(1):830–836. doi: 10.1128/jvi.72.1.830-836.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernanz-Falcón P, et al. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat Immunol. 2004;5(2):216–223. doi: 10.1038/ni1027. [DOI] [PubMed] [Google Scholar]
- 19.Percherancier Y, et al. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem. 2005;280(11):9895–9903. doi: 10.1074/jbc.M411151200. [DOI] [PubMed] [Google Scholar]
- 20.Sohy D, et al. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J Biol Chem. 2009;284(45):31270–31279. doi: 10.1074/jbc.M109.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrero-Villar M, et al. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J Cell Sci. 2009;122(Pt 1):103–113. doi: 10.1242/jcs.035873. [DOI] [PubMed] [Google Scholar]
- 22.Lapham CK, et al. Evidence for cell-surface association between fusin and the CD4-gp120 complex in human cell lines. Science. 1996;274(5287):602–605. doi: 10.1126/science.274.5287.602. [DOI] [PubMed] [Google Scholar]
- 23.Ugolini S, et al. HIV-1 gp120 induces an association between CD4 and the chemokine receptor CXCR4. J Immunol. 1997;159(6):3000–3008. [PubMed] [Google Scholar]
- 24.Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1(3):1278–1286. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carriba P, et al. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat Methods. 2008;5(8):727–733. doi: 10.1038/nmeth.1229. [DOI] [PubMed] [Google Scholar]
- 26.Martínez Muñoz L, et al. Dynamic regulation of CXCR1 and CXCR2 homo- and heterodimers. J Immunol. 2009;183(11):7337–7346. doi: 10.4049/jimmunol.0901802. [DOI] [PubMed] [Google Scholar]
- 27.Yoder A, et al. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell. 2008;134(5):782–792. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balabanian K, et al. CXCR4-tropic HIV-1 envelope glycoprotein functions as a viral chemokine in unstimulated primary CD4+ T lymphocytes. J Immunol. 2004;173(12):7150–7160. doi: 10.4049/jimmunol.173.12.7150. [DOI] [PubMed] [Google Scholar]
- 29.Nishita M, Aizawa H, Mizuno K. Stromal cell-derived factor 1alpha activates LIM kinase 1 and induces cofilin phosphorylation for T-cell chemotaxis. Mol Cell Biol. 2002;22(3):774–783. doi: 10.1128/MCB.22.3.774-783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasiliver-Shamis G, et al. Human immunodeficiency virus type 1 envelope gp120 induces a stop signal and virological synapse formation in noninfected CD4+ T cells. J Virol. 2008;82(19):9445–9457. doi: 10.1128/JVI.00835-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hioe CE, et al. HIV envelope gp120 activates LFA-1 on CD4 T-lymphocytes and increases cell susceptibility to LFA-1-targeting leukotoxin (LtxA) PLoS ONE. 2011;6(8):e23202. doi: 10.1371/journal.pone.0023202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vorster PJ, et al. LIM kinase 1 modulates cortical actin and CXCR4 cycling and is activated by HIV-1 to initiate viral infection. J Biol Chem. 2011;286(14):12554–12564. doi: 10.1074/jbc.M110.182238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vega B, et al. Technical advance: Surface plasmon resonance-based analysis of CXCL12 binding using immobilized lentiviral particles. J Leukoc Biol. 2011;90(2):399–408. doi: 10.1189/jlb.1010565. [DOI] [PubMed] [Google Scholar]
- 34.Kozak SL, Kuhmann SE, Platt EJ, Kabat D. Roles of CD4 and coreceptors in binding, endocytosis, and proteolysis of gp120 envelope glycoproteins derived from human immunodeficiency virus type 1. J Biol Chem. 1999;274(33):23499–23507. doi: 10.1074/jbc.274.33.23499. [DOI] [PubMed] [Google Scholar]
- 35.Thelen M, Muñoz LM, Rodríguez-Frade JM, Mellado M. Chemokine receptor oligomerization: Functional considerations. Curr Opin Pharmacol. 2010;10(1):38–43. doi: 10.1016/j.coph.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 36.Del Real G, et al. Blocking of HIV-1 infection by targeting CD4 to nonraft membrane domains. J Exp Med. 2002;196(3):293–301. doi: 10.1084/jem.20020308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, He L, Combs CA, Roderiquez G, Norcross MA. Dimerization of CXCR4 in living malignant cells: Control of cell migration by a synthetic peptide that reduces homologous CXCR4 interactions. Mol Cancer Ther. 2006;5(10):2474–2483. doi: 10.1158/1535-7163.MCT-05-0261. [DOI] [PubMed] [Google Scholar]
- 38.Wilson S, Wilkinson G, Milligan G. The CXCR1 and CXCR2 receptors form constitutive homo- and heterodimers selectively and with equal apparent affinities. J Biol Chem. 2005;280(31):28663–28674. doi: 10.1074/jbc.M413475200. [DOI] [PubMed] [Google Scholar]
- 39.Mellado M, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001;20(10):2497–2507. doi: 10.1093/emboj/20.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Contento RL, et al. CXCR4-CCR5: A couple modulating T cell functions. Proc Natl Acad Sci USA. 2008;105(29):10101–10106. doi: 10.1073/pnas.0804286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muñoz LM, et al. Receptor oligomerization: A pivotal mechanism for regulating chemokine function. Pharmacol Ther. 2011;131(3):351–358. doi: 10.1016/j.pharmthera.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 42.Sierro F, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci USA. 2007;104(37):14759–14764. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113(24):6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- 44.Isik N, Hereld D, Jin T. Fluorescence resonance energy transfer imaging reveals that chemokine-binding modulates heterodimers of CXCR4 and CCR5 receptors. PLoS ONE. 2008;3(10):e3424. doi: 10.1371/journal.pone.0003424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferré S, et al. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5(3):131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Audet N, et al. Bioluminescence resonance energy transfer assays reveal ligand-specific conformational changes within preformed signaling complexes containing delta-opioid receptors and heterotrimeric G proteins. J Biol Chem. 2008;283(22):15078–15088. doi: 10.1074/jbc.M707941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M. Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem. 2002;277(47):44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- 48.Adachi A, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59(2):284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399(6737):697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barroso R, et al. EBI2 regulates CXCL13-mediated responses by heterodimerization with CXCR5. FASEB J. 2012;26(12):4841–4854. doi: 10.1096/fj.12-208876. [DOI] [PubMed] [Google Scholar]
- 51.Décaillot FM, et al. CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. J Biol Chem. 2011;286(37):32188–32197. doi: 10.1074/jbc.M111.277038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baleux F, et al. A synthetic CD4-heparan sulfate glycoconjugate inhibits CCR5 and CXCR4 HIV-1 attachment and entry. Nat Chem Biol. 2009;5(10):743–748. doi: 10.1038/nchembio.207. [DOI] [PubMed] [Google Scholar]
- 53.Toth PT, Ren D, Miller RJ. Regulation of CXCR4 receptor dimerization by the chemokine SDF-1alpha and the HIV-1 coat protein gp120: A fluorescence resonance energy transfer (FRET) study. J Pharmacol Exp Ther. 2004;310(1):8–17. doi: 10.1124/jpet.103.064956. [DOI] [PubMed] [Google Scholar]
- 54.Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA. 1997;94(5):1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gosselin A, et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J Immunol. 2010;184(3):1604–1616. doi: 10.4049/jimmunol.0903058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Alvarez R, Roderiquez G, Guan E, Norcross MA. Constitutive association of cell surface CCR5 and CXCR4 in the presence of CD4. J Cell Biochem. 2004;93(4):753–760. doi: 10.1002/jcb.20161. [DOI] [PubMed] [Google Scholar]
- 57.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72(4):2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trkola A, et al. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc Natl Acad Sci USA. 2002;99(1):395–400. doi: 10.1073/pnas.012519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Margolis L, Shattock R. Selective transmission of CCR5-utilizing HIV-1: The ‘gatekeeper’ problem resolved? Nat Rev Microbiol. 2006;4(4):312–317. doi: 10.1038/nrmicro1387. [DOI] [PubMed] [Google Scholar]
- 60.Patterson BK, et al. Repertoire of chemokine receptor expression in the female genital tract: Implications for human immunodeficiency virus transmission. Am J Pathol. 1998;153(2):481–490. doi: 10.1016/S0002-9440(10)65591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller CJ, Shattock RJ. Target cells in vaginal HIV transmission. Microbes Infect. 2003;5(1):59–67. doi: 10.1016/s1286-4579(02)00056-4. [DOI] [PubMed] [Google Scholar]
- 62.Veazey RS, Marx PA, Lackner AA. Vaginal CD4+ T cells express high levels of CCR5 and are rapidly depleted in simian immunodeficiency virus infection. J Infect Dis. 2003;187(5):769–776. doi: 10.1086/368386. [DOI] [PubMed] [Google Scholar]
- 63.Poles MA, Elliott J, Taing P, Anton PA, Chen IS. A preponderance of CCR5(+) CXCR4(+) mononuclear cells enhances gastrointestinal mucosal susceptibility to human immunodeficiency virus type 1 infection. J Virol. 2001;75(18):8390–8399. doi: 10.1128/JVI.75.18.8390-8399.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galli G, et al. Th1 and th2 responses, HIV-1 coreceptors, and HIV-1 infection. J Biol Regul Homeost Agents. 2001;15(3):308–313. [PubMed] [Google Scholar]
- 65.Berkowitz RD, Alexander S, McCune JM. Causal relationships between HIV-1 coreceptor utilization, tropism, and pathogenesis in human thymus. AIDS Res Hum Retroviruses. 2000;16(11):1039–1045. doi: 10.1089/08892220050075291. [DOI] [PubMed] [Google Scholar]
- 66.Motulsky H, Christopoulos A. Fitting Models to Biological Data using Linear and Nonlinear Regression. A Practical Guide to Curve Fitting. New York: Oxford Univ Press; 2004. [Google Scholar]
- 67.Poncelet P, George F, Lavabre-Bertrand T. Immunological detection of membrane bound antigens and receptors. In: Masseyesff R, Staines N, Albert W, editors. Methods of Immunological Analysis. Weinheim, Germany: Wiley; 1993. pp. 388–418. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.