

Significance
Toll-like receptors (TLRs) are key innate immune receptors that sense pathogen- or danger-associated molecules. The members of the TLR family can be classified into two groups based on their subcellular localization patterns—cell surface vs. endolysosomes—but how individual TLR is targeted to its destination is largely unknown. UNC93B1 was regarded to specifically bind the endolysosomal TLRs in the endoplasmic reticulum and deliver them to endolysosomes with no role for the surface-localized TLRs. In this study, we demonstrate that TLR5, a cell surface receptor for bacterial protein flagellin, also binds UNC93B1 and requires it for plasma membrane localization and signaling, thus revealing a previously unappreciated role of UNC93B1 in the regulation of the TLR family.
Keywords: Toll-like receptor trafficking, 3d
Abstract
The proper trafficking and localization of Toll-like receptors (TLRs) are important for specific ligand recognition and efficient signal transduction. The TLRs sensing bacterial membrane components are expressed on the cell surface and recruit signaling adaptors to the plasma membrane upon stimulation. On the contrary, the nucleotide-sensing TLRs are mostly found inside cells and signal from the endolysosomes in an acidic pH-dependent manner. Trafficking of the nucleotide-sensing TLRs from the endoplasmic reticulum to the endolysosomes strictly depends on UNC93B1, and their signaling is completely abolished in the 3d mutant mice bearing the H412R mutation of UNC93B1. In contrast, UNC93B1 was considered to have no role for the cell surface-localized TLRs and signaling via TLR1, TLR2, TLR4, and TLR6 is normal in the 3d mice. Unexpectedly, we discovered that TLR5, a cell surface receptor for bacterial protein flagellin, also requires UNC93B1 for plasma membrane localization and signaling. TLR5 physically interacts with UNC93B1, and the cells from the 3d or UNC93B1-deficient mice not only lack TLR5 at the plasma membrane but also fail to secret cytokines and to up-regulate costimulatory molecules upon flagellin stimulation, demonstrating the essential role of UNC93B1 in TLR5 signaling. Our study reveals that the role of UNC93B1 is not limited to the TLRs signaling from the endolysosomes and compels the further probing of the mechanisms underlying the UNC93B1-assisted differential targeting of TLRs.
Toll-like receptors (TLRs) sense unique microbial structures or host-derived molecules released from stressed or dying cells to initiate the innate immune responses (1). TLRs are composed of three domains: the leucine-rich repeat (LRR) domain responsible for ligand binding, a single transmembrane domain, and the cytoplasmic Toll/IL-1 receptor homology domain by which TLRs recruit adaptor molecules for downstream signal transduction. Activated TLRs stimulate the NF-κB, MAPK, and IFN regulatory factor pathways, leading to the expression of diverse inflammatory cytokines, chemokines, and type I interferons. TLRs also activate antigen presenting cells to induce costimulatory molecules and coordinate various aspects of adaptive immune responses (2).
The members of the TLR family can be classified into two groups based on their subcellular localization patterns (3–5). TLR1, TLR2, TLR4, and TLR6, which mainly recognize the components of bacterial cell membrane, are located on the cell surface and initiate signaling thereat. In contrast, the nucleotide-sensing TLRs such as TLR3, TLR7, TLR8, TLR9, and TLR13 are largely found in endolysosomes and require an acidic environment for their efficient signaling. Additionally, TLR11 and TLR12, the sensors for Toxoplasma protein profilin, are also expressed inside cells and transmit signals in an acidic pH-dependent manner (6–8). All the intracellular TLRs commonly bind to a multispanning membrane protein UNC93B1, which is required for their proper localization and signaling (6–13). One missense mutation (H412R) of UNC93B1, found in a chemically mutagenized mouse strain called 3d, hinders binding of UNC93B1 with TLRs and prevents their exit from the endoplasmic reticulum (ER) (9–11). Consequently, signaling by all endosomal TLRs is abolished in the cells from 3d mice. In contrast, trafficking and signaling of the cell surface-localized TLRs such as TLR2 and TLR4 are not affected by the UNC93B1 mutation (9, 11).
The proper localization of TLRs is critical not only for efficient signaling but also for preventing undesirable receptor hyperactivation (14, 15). Especially, sequestration of the nucleotide-sensing TLRs in endolysosomes significantly contributes to attenuating the immune stimulation by host-derived nucleotides abundant in the extracellular spaces (14). Structural discrimination of microbial vs. mammalian nucleotides is not straightforward, and a mutant TLR9 protein, engineered to artificially localize at the plasma membrane, responds to mammalian DNA as well as the CpG oligonucleotides mimicking bacterial DNA. As a result, mice expressing such mutant TLR9 succumb to systemic autoinflammation and die prematurely (15). Therefore, regulatory mechanisms for localization and trafficking of TLRs need to be tightly controlled.
TLR5 recognizes flagellin, the major protein subunit of bacterial flagellum, and functions as a critical innate sensor for flagellated bacteria in all mucous organs (16–18). TLR5 plays an important role in intestinal homeostasis mediating the immune adaptation to symbiotic microflora as well as defense against pathogenic bacterial infection (19–21). In addition, systemic injection of flagellin confers protection against ionizing radiation in a TLR5-dependent manner, implying that TLR5 agonism might be clinically used for radioprotection (22). TLR5 overexpressed in the intestinal epithelial cells was exclusively found on the basolateral surface, accounting for the selective induction of proinflammatory cytokine by basolateral but not by apical flagellin (17). Also, we recently demonstrated that endogenous TLR5 is expressed at the cell surface of mouse neutrophils, monocytes, and dendritic cells (DCs) in a TLR-specific chaperone PRAT4A-dependnet manner (23). However, other regulatory mechanisms for the localization of TLR5 at the plasma membrane are unknown. Here, we show that UNC93B1 binds to TLR5, travels to the plasma membrane with the receptor, and is required for flagellin-induced signaling at the cell surface.
Results
TLR5 Binds to UNC93B1.
Previously, we showed that acidic amino acid residues located between the LRR domain and the transmembrane segment of TLR3 and TLR9 play an important role for the receptor interaction with UNC93B1 (24). Moreover, acidic residues were commonly found in the same region of other UNC93B1-binding endosomal TLRs, including TLR7 and TLR8, but not in the surface-localized TLRs, TLR2 or TLR4. Interestingly, we noticed that TLR5, a cell surface TLR and thus the interaction of which with UNC93B1 was not expected, also contains four acidic residues (DEEE) in the same region (Fig. 1A). Therefore, we tested if TLR5 interacts with UNC93B1 by using HEK 293T cells transiently expressing UNC93B1 and TLRs. UNC93B1 was coimmunoprecipitated with TLR5 as well as with TLR9, but not with TLR4, indicating that TLR5 binds to UNC93B1 (Fig. 1B). Moreover, in RAW264.7 mouse macrophage cells stably expressing TLR5-myc, endogenous UNC93B1 was coimmunoprecipitated with TLR5, confirming the interaction between TLR5 and UNC93B1 (Fig. 1C).
Fig. 1.

TLR5 binds to UNC93B1. (A) Amino acid sequence alignment of the putative transmembrane and surrounding regions of human TLRs. The alignment starts at the conserved cysteine residue (in bold type) located at the end of the LRR domain and the corresponding amino acid residue number of each TLR is indicated. The putative membrane-inserted sequences are predicted by using ΔG prediction server version 1.0 (http://dgpred.cbr.su.se/) and represented by light blue shading (33). Acidic amino acid residues are highlighted in red. (B) UNC93B1-HA and TLR-myc were transiently expressed in HEK 293T cells. TLRs were immunoprecipitated from the cell lysate and detected by immunoblotting with anti-myc antibodies (Bottom). The levels of the coimmunoprecipitated UNC93B1 (Top) and the total UNC93B1 in the input lysate (Middle) were probed with an anti-HA antibody. Data shown are representative of three independent experiments. (C) The lysate of RAW cells stably expressing TLR5-myc was subjected to immunoprecipitation with anti-GFP or anti-myc antibody. Endogenous UNC93B1 bound to TLR5-myc was detected by immunoblotting with anti-UNC93B1 antibody (Top). Input lysate was probed for endogenous UNC93B1 (Middle) and TLR5-myc (Bottom) to check expression levels. Data shown are representative of three independent experiments.
UNC93B1 Is Required for the Cell Surface Localization of TLR5.
Next, we tested if UNC93B1 is required for the localization of TLR5 at the plasma membrane. The level of endogenous TLR5 at the cell surface of J774 macrophage line was measured by flow cytometry by using a monoclonal TLR5 antibody (23). We could clearly detect TLR5 at the cell surface of control J774 cells, but not when UNC93B1 was knocked down with a specific shRNA (Fig. 2A and Fig. S1A). As expected, knockdown of UNC93B1 did not affect the cell surface expression of TLR4/MD2 or CD14. We also found that knockdown of UNC93B1 in Ba/F3 cells stably expressing TLR5-flag (Ba/F3:TLR5-flag) prevented the surface expression of TLR5 without affecting the total expression level of TLR5 or the cell surface level of endogenous TLR2 (Fig. S1 B and C). Furthermore, we observed that overexpression of WT but not H412R mutant UNC93B1 increased cell surface levels of TLR5 (Fig. S1D).
Fig. 2.

UNC93B1 is essential for the cell surface expression of TLR5. (A) The cell surface levels of TLR5, TLR4/MD-2, and CD14 in J774 cells expressing control or Unc93B1 shRNA were analyzed (open histogram). Shaded histograms represent control staining with streptavidin-PE alone. Data shown are representative of three independent experiments. (B and C) Blood leukocytes (B) and splenic DCs (C) from WT, 3d, and TLR5−/− mice were subjected to analysis for cell surface expression of TLR5. The CD11b+ fraction in blood leukocytes was subdivided into Ly6CintLy6G+ (R1), Ly6ClowLy6G− (R2), Ly6CintLy6G− (R3), and Ly6ChiLy6G− (R4) populations. In C, CD11cintPDCA-1+ pDCs and CD11chiPDCA-1− cDCs were gated from whole splenocytes and the cDCs were subdivided into CD8α+CD4− (CD8α+ cDC), CD8α−CD4+ (CD4+ cDC), and CD8α−CD4− (DN cDC). Open histograms represent the cell surface TLR5 levels and shaded histograms represent control staining with streptavidin-PE alone. Data shown are representative of three independent experiments. (D) Leukocytes from small intestinal lamina propria of WT and 3d mice were subjected to analysis for cell surface expression of TLR5. The surface level of TLR5 in CD11bhighCD11chigh LP-DCs (R1) and CD11bhighCD11cint eosinophils (R2) are shown in open histograms. Shaded histograms represent control staining with streptavidin-PE alone. Data shown are representative of two independent experiments.
In mouse blood, TLR5 is mainly expressed in neutrophils and CD11bhighLy6Chi classical monocytes (23). In contrast to WT cells, we found that the cell surface expression of TLR5 was absent in neutrophils and classical monocytes from 3d mice expressing the mutant UNC93B1 protein (Fig. 2B). Additionally, we verified that the normal cell surface expression of TLR5 was lost in other cell types of 3d mice, including splenic plasmacytoid DCs (pDCs), CD4+ conventional DCs (cDCs), CD4−CD8− cDCs, and small intestine lamina propria DCs (LP-DCs; Fig. 2 C and D). These data demonstrate that the functional UNC93B1 protein is required for the plasma membrane localization of TLR5.
The role of UNC93B1 in the cell surface localization of TLR5 was confirmed by confocal microscopy. When TLR5-GFP was expressed in WT bone marrow-derived DCs (BMDCs), a large proportion of TLR5-GFP was colocalized with cholera toxin B, a marker for the plasma membrane. In contrast, TLR5-GFP did not colocalize with cholera toxin B in BMDCs from the UNC93B1-deficient or 3d mice and was exclusively found inside the cells (Fig. 3A and Figs. S2 and S3 A and B). The failure of TLR5 to localize at the cell surface of the UNC93B1-deficient BMDCs was corrected by expression of UNC93B1-cherry (Fig. 3B) Similarly, coexpression of WT UNC93B1 resulted in the localization of TLR5-GFP at the plasma membrane in HEK 293T cells that normally have a limited amount of endogenous UNC93B1, whereas TLR5-GFP was almost exclusively found in the ER without the exogenous UNC93B1 (Fig. S3C) Coexpression of the H412R mutant UNC93B1 failed to induce the cell surface localization of TLR5-GFP and the receptor was retained in the ER, indicating that WT UNC93B1 is required for exit of TLR5 from the ER. Accordingly, TLR5 in the UNC93B1 knockdown cells did not acquire the complex type of oligosaccharides in the Golgi apparatus and remained sensitive to digestion with endoglycosidase H (Fig. S2E).
Fig. 3.

Localization of TLR5 at the plasma membrane requires UNC93B1. (A) WT or UNC93B1-deficient (KO) BMDCs expressing TLR5-GFP were stained with Alexa Fluor 555-conjugated cholera toxin B. Data shown are representative of two independent experiments. (B) TLR5-GFP with or without UNC93B1-cherry were expressed in WT or KO BMDCs. Data shown are representative of three independent experiments. (Scale bar, 10 μm.)
Previously, we reported that UNC93B1 escorts TLR9 to endolysosomes and showed the colocalization of WT UNC93B1-cherry with TLR9-GFP in endolysosomes (11). The requirement of UNC93B1 for the localization of TLR5 at the plasma membrane raises a possibility that UNC93B1 might travel to the plasma membrane together with TLR5. Indeed, we found that UNC93B1-cherry colocalized with TLR5-GFP in HEK 293T cells and BMDCs (Fig. 3B and Fig. S4A). The surface localization of UNC93B1-cherry was evident only in the cells overexpressing TLR5. To confirm the colocalization of UNC93B1 with TLR5 at the plasma membrane, we adopted the bimolecular fluorescent complementation assay by using the Venus fluorescent protein. The N-terminal half of Venus (VN) was fused to the C-terminal end of TLRs to generate the TLR-VN fusion proteins, and the C-terminal half of Venus (VC) was fused to the N- or C-terminal end of UNC93B1 to generate VC-UNC93B1 or UNC93B1-VC, respectively (Fig. S4B). We also generated the TLR-VC fusion proteins in which VC was fused to the C-terminal end of TLRs. When TLR-VN and TLR-VC were coexpressed, cells were lit up with the Venus fluorescence signal. In the cells coexpressing TLR9-VN and TLR9-VC, the fluorescence signal was detected only inside the cells, whereas the cells with the combination of TLR4-VN and TLR4-VC or TLR5-VN and TLR5-VC showed significant fluorescent signal at the cell surface, indicating the localization of the receptor dimers on the plasma membrane. Similarly, robust fluorescent signals were detected at the surface of the cells expressing TLR5-VN together with UNC93B1-VC or VC-UNC93B1. In contrast, expression of TLR9-VN with UNC93B1-VC or VC-UNC93B1 produced the fluorescent signal exclusively inside the cells. As expected, no fluorescent signal was detected in the cells with combination of TLR4-VN with UNC93B1-VC or VC-UNC93B1. Taken together, these data strongly support the localization of TLR5-bound UNC93B1 at the plasma membrane.
TLR5-GFP signal was also visible inside the WT BMDCs but did not colocalize with the endolysosomal marker protein CD63-cherry (Fig. S3B). Moreover, bafilomycin A, an inhibitor for endolysosomal V-type ATPase, did not affect flagellin-induced TNF-α production, whereas it severely abrogated TNF-α production by R848 and CpG, agonists for endolysosomal TLR7 and TLR9, respectively, (Fig. S5). These data suggest that TLR5 most likely recognizes extracellular flagellin and signals from the cell surface unlike the other UNC93B1-dependent TLRs, which signal from the endolysosomes in a pH-dependent manner. Therefore, we predicted that the lack of cell surface TLR5 in the UNC93B1-deficient or mutant cells would result in the loss of TLR5 signaling.
UNC93B1 Is Required for Signaling of TLR5.
We first measured the flagellin-induced cytokine secretion from J774 cells expressing UNC93B1-specific shRNA. Unlike the control cells, The UNC93B1 knockdown cells did not produce IL-6 or granulocyte-colony stimulating factor (G-CSF) in response to flagellin, as they did not respond to the TLR9 agonist CpG and the TLR7 agonist loxoribine (Fig. 4A). In addition, flagellin failed to up-regulate the expression of CD40 in the UNC93B1 knockdown cells (Fig. 4B). These data indicate that UNC93B1 is required for flagellin-induced TLR5 signaling.
Fig. 4.
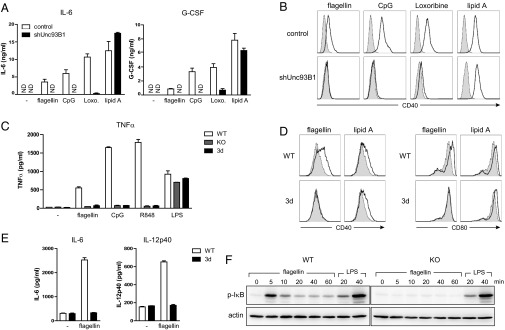
UNC93B1 is essential for TLR5-mediated signaling in vitro. (A) J774 cells expressing control or Unc93B1 shRNA were stimulated with flagellin (100 ng/mL), CpG (ODN1668, 100 nM), loxoribine (100 μM), or lipid A (100 ng/mL) for 20 h. Production of IL-6 and G-CSF was measured by ELISA. The data represent mean ± SD of triplicate samples. ND, not detected. Data shown are representative of three independent experiments. (B) J774 cells treated as in A were analyzed for surface expression of CD40 (open histogram). Shaded histograms represent levels of CD40 in unstimulated cells. Data shown are representative of three independent experiments. (C) Splenic DCs from WT, UNC93B1-deficinent, or 3d mice were stimulated with flagellin (100 ng/mL), CpG (ODN1826; 1 μM), R848 (1 μM), or LPS (100 ng/mL) for 4 h, and levels of TNF-α were measured by ELISA (mean ± SD). Data shown are representative of two independent experiments. (D) Splenocytes from WT or 3d mice were stimulated with flagellin (100 ng/mL) or lipid A (100 ng/mL) for 20 h. Open histograms represent expression levels of CD40 (Left) or CD80 (Right) on CD11chiPDCA-1−CD8α−-gated cells (CD8α− cDCs). Shaded histograms represent the levels of CD40 or CD80 in unstimulated cells. (E) Small intestinal LP-DCs from WT or 3d mice were stimulated with 1 μg/mL of flagellin or left untreated for 24 h, and levels of IL-6 (Left) or IL-12p40 (Right) were measured by ELISA. The data represent mean ± SD of triplicate samples. Data shown are representative of three independent experiments. (F) WT or UNC93B1-deficient BMDCs were stimulated with flagellin (100 ng/mL) or LPS (100 ng/mL). The phosphorylated IκB (Upper) and actin (Lower) were detected by immunoblotting. Data shown are representative of two independent experiments.
We confirmed the importance of UNC93B1 for TLR5 signaling by using primary cells from the UNC93B1-deficient or 3d mice. Flagellin induced secretion of TNF-α as well as up-regulation of CD40 and CD86 in the splenic DCs from WT but not from the UNC93B1-deficient or 3d mice (Fig. 4 C and D). BMDCs hardly express TLR5 and produce little TNF-α upon stimulation with flagellin. However, when we expressed TLR5-GFP in WT BMDCs, flagellin strongly induced the production of TNF-α (Fig. S6A). In contrast, BMDCs from the UNC93B1-deficient mice did not secrete TNF-α upon flagellin stimulation even with overexpression of TLR5-GFP. We also found that flagellin stimulation led to the secretion of IL-6 and IL-12p40 from LP-DCs purified from WT but not from 3d mice (Fig. 4E). Collectively, UNC93B1 was required for the TLR5-mediated cytokine secretion as well as the costimulatory molecule induction in all cells tested.
We also found that flagellin treatment resulted in the phosphorylation of IκB only in WT but not in UNC93B1-deficient BMDCs, showing that the lack of UNC93B1 and the consequent receptor mislocalization prevented TLR5-mediated activation of the NF-κB pathway (Fig. 4F). This was further confirmed by the finding that the flagellin-induced expression of NF-κB–GFP reporter gene was severely attenuated in the cells expressing shRNA for Unc93B1 (Fig. S6B).
To test whether UNC93B1 is physiologically important for TLR5 signaling in vivo, we intraperitoneally injected 1 μg of flagellin into WT or UNC93B1-deficient mice and measured the serum levels of IL-6 and IL-12p40 as well as the levels of CD40, CD80, and CD86 in splenic DCs. Consistent with the in vitro experimental results demonstrating the importance of UNC93B1 in TLR5 signaling, the serum concentration of IL-6 and IL-12p40 significantly increased only in WT but not in the UNC93B1-deficient mice upon flagellin injection (Fig. 5A). Induction of CD40, CD80, and CD86 was also observed only in the WT mice (Fig. 5B).
Fig. 5.

UNC93B1 is essential for TLR5-mediated signaling in vivo. (A and B) WT and UNC93B1-deficient mice (n = 3 each) were injected i.p. with 1 μg of flagellin. (A) The serum levels of IL-6 and IL-12p40 were measured by ELISA (mean ± SD). (B) Six hours later, splenocytes were collected and the levels of CD40, CD80, and CD86 in CD11chigh DCs were analyzed by flow cytometry.
TLR5 is one of the key innate immune receptors operating upon Pseudomonas aeruginosa infection (18). Accordingly, WT splenic DC produced less IL-12p40 when stimulated with a flagellin-deficient mutant strain (ΔfliC) compared with WT P. aeruginosa (Fig. S7). In contrast, TLR5- and UNC93B1-deficient cells responded equally to WT and ΔfliC strains, confirming the role of TLR5 and UNC93B1 in recognition of P. aeruginosa flagellin.
Acidic Amino Acid Residues in the Juxtamembrane Region of TLR5 Are Important for Binding to UNC93B1.
We tested if the acidic amino acid residues in the juxtamembrane region of TLR5 participate in the interaction with UNC93B1 as they did for TLR3 and TLR9 (24). For this, we replaced the DEEE sequence (amino acid residues 630∼633) of TLR5-GFP with STTT to generate TLR5-GFP (DE/ST). This mutant TLR5 was highly impaired in binding to UNC93B1 (Fig. 6A). When expressed in BMDCs, the mutant TLR5-GFP (DE/ST) was barely detected at the plasma membrane compared with WT TLR5-GFP (Fig. 6B). Furthermore, BMDCs expressing the mutant TLR5-GFP (DE/ST) showed the reduction of TNF-α production upon flagellin stimulation compared with the cells with WT TLR5-GFP (Fig. 6C). Taken together, the acidic residues in the juxtamembrane region of TLR5 are involved in binding to UNC93B1; thus, the replacement of those residues to noncharged amino acids hampers trafficking of TLR5 to the plasma membrane and the subsequent signaling.
Fig. 6.

Acidic amino acid residues in the juxtamembrane region of TLR5 are important for UNC93B1 binding and signaling. (A) TLR5 was immunoprecipitated from RAW cells stably expressing UNC93B1-HA with TLR5-GFP (WT or DE/ST). Coimmunoprecipitated UNC93B1-HA (Top) as well as UNC93B1-HA (Middle) and TLR5-GFP (Bottom) in the input lysate were detected by immunoblotting. The data shown are representative of three independent experiments. (B) BMDCs expressing WT or DE/ST mutant TLR5-GFP were imaged. (C) BMDCs expressing GFP or TLR5-GFP (WT or DE/ST mutant) were stimulated with flagellin (100 ng/mL) or LPS (100 ng/mL) for 4 h and the intracellular TNF-α levels in the GFP-positive cells were measured (open histograms). Data shown are representative of three independent experiments. Shaded histograms represent the levels of TNF-α in unstimulated cells.
Discussion
The subcellular destination of a TLR greatly influences the varieties of available agonists for the receptor, its ligand binding affinities, and the downstream signaling modes, and thus the tight regulation of the intracellular TLR trafficking is crucial for the optimal receptor activation. UNC93B1 was initially identified as an essential factor that specifically binds and delivers the nucleotide-sensing TLRs to endolysosomes. More recently, protozoan profilin-sensing TLR11 and TLR12 were also shown to be intracellular TLRs and to depend on UNC93B1 for their endosomal localization and signaling. Because trafficking and signaling of the cell surface-disposed TLRs such as TLR1, TLR2, TLR4, and TLR6 are independent of UNC93B1, it was generally believed that the role of UNC93B1 is limited to the intracellular TLRs. However, in this study, we found that TLR5, a cell surface-localized TLR, is another client of UNC93B1, and demonstrated that TLR5 relies on UNC93B1 for its localization and signaling at the plasma membrane. Therefore, the role of UNC93B1 is not restricted to the TLRs that initiate signaling from the endolysosomes.
Notably, we found that, in addition to its expected localization in the ER and endolysosomes, UNC93B1 accumulated at the surface of cells expressing TLR5. From the bimolecular fluorescent complementation assay using the split Venus proteins fused to UNC93B1 and TLR5, we observed that the surface-localized UNC93B1 remained bound to TLR5 at the plasma membrane. Together with our previous finding that UNC93B1 travels to endolysosomes along with TLR9, it shows that the interaction between UNC93B1 and TLRs established in the ER is maintained throughout the intracellular trafficking pathways to the final destinations.
TLR9 was recently shown to transit through the cell surface en route to the endolysosomes (13). UNC93B1 mediates not only the exit of TLR9 from the ER but also controls its internalization from the plasma membrane by recruiting the clathrin adaptor AP-2. In the AP-2 knockdown cells or cells expressing the UNC93B1 mutant protein defective in AP-2 binding, TLR9 accumulated at the plasma membrane and CpG-induced signaling was blunted. In such cells, however, trafficking and signaling of other UNC93B1-dependent TLRs including TLR7, TLR11, TLR12, and TLR13 were not altered, indicating that post-ER trafficking of these TLRs is differentially regulated from that of TLR9. Interestingly, TLR9 was reported to be present on the cell surface of intestinal epithelial cells, splenic DCs, and neutrophils (25–27). Similarly, TLR3 can be found on the surface of epithelial cells and fibroblasts even though it is exclusively located inside the myeloid cells (28, 29). These observations raise the possibility that TLR3 may follow a similar trafficking pathway as TLR9 and that the internalization efficiency of TLR3 and TLR9 varies depending on cell type. In case of TLR5, so far, all published reports as well as our own data support that it remains on the cell surface at steady state after the UNC93B1-dependent trafficking to the plasma membrane (17, 23).
How individual UNC93B1-dependent TLRs are targeted to different subcellular organelles is in need of further investigation. Mutational studies of UNC93B1 revealed that TLRs have differential affinities for UNC93B1, and consequently UNC93B1 promotes the preferential exit of TLR9 from the ER compared with TLR7 (30). After leaving the ER, accumulating data suggest the initial sorting of UNC93B1-bound TLRs in the trans-Golgi network. TLR5, TLR9, and possibly TLR3 seem to travel to the cell surface via the traditional anterograde transport pathway and are sorted again at the plasma membrane. TLR5 stays at the cell surface whereas TLR9 and TLR3 mostly become internalized into endolysosomes. In contrast, the other UNC93B1-dependent TLRs seem to directly travel to endolysosomes from the trans-Golgi network because their trafficking is not affected by the mutation of UNC93B1 causing the loss of AP-2 binding (13). Alternatively, they might also travel to the plasma membrane first, but internalize to endolysosomes via an AP-2-independent manner or directly bind AP-2 instead of recruiting AP-2 via UNC93B1.
TLR5 was proposed to function as an endocytic receptor for flagellin (31). Thus, it would be interesting to investigate the changes of TLR5 localization upon flagellin binding and the role of UNC93B1 in such processes. In BMDCs, a large proportion of TLR9 was found in the ER at steady state and moved to endolysosomes upon CpG stimulation, implying the active regulation of UNC93B1-mediated TLR trafficking (11, 32). Studying the regulatory mechanisms of UNC93B1 in various stimulatory conditions and different cell types would help to elucidate how intracellular trafficking of individual TLRs is controlled for efficient and specific detection of pathogens while avoiding overactivation by innocuous self molecules.
Materials and Methods
Immunoprecipitation and immunoblotting, preparation of BMDCs, and intracellular TNF assay were performed as previously described (24). The cell surface levels of TLR2, TLR5, TLR4/MD-2, and CD14 were analyzed by flow cytometry by using biotinylated anti-TLR2 (CB225), anti-TLR5 (ACT5), anti-TLR4/MD-2 (MTS510), and anti-CD14 (Sa2-8) antibodies, respectively. More information is provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Seung-Woo Lee (Pohang University of Science and Technology) and the members of the Hyehwa Forum for helpful discussions. The 3d mice and vectors for the bimolecular fluorescent complementation assay were provided by Koichi Tabeta (Niigata University) and Michael Frohman (Stony Brook University), respectively. Pseudomonas aeruginosa wild type PAO1 and fliC-deficient strains were kindly provided by Yong Song Gho (Pohang University of Science and Technology). This work was supported by National Research Foundation of Korea Grant 2011-0016801; BK21 Plus Grant 10Z20130012243; the Institute for Basic Science; Pohang University of Science and Technology Basic Science Research Institute grant; a contract research fund from the Ministry of Education, Culture, Sports, Science and Technology for Program of Japan Initiative for Global Research Network on Infectious Diseases; Grant-in-Aid for Scientific Research (B) no. 25860354; and Grant-in-Aid for Scientific Research on Innovative Areas no. 21117002.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. H.L.P. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322838111/-/DCSupplemental.
References
- 1.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat Immunol. 2010;11(5):373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 2.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327(5963):291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: Regulation through compartmentalization. Nat Rev Immunol. 2009;9(8):535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32(3):305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Saitoh S, Miyake K. Regulatory molecules required for nucleotide-sensing Toll-like receptors. Immunol Rev. 2009;227(1):32–43. doi: 10.1111/j.1600-065X.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- 6.Pifer R, Benson A, Sturge CR, Yarovinsky F. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J Biol Chem. 2011;286(5):3307–3314. doi: 10.1074/jbc.M110.171025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koblansky AA, et al. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity. 2013;38(1):119–130. doi: 10.1016/j.immuni.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrade WA, et al. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe. 2013;13(1):42–53. doi: 10.1016/j.chom.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabeta K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7(2):156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 10.Brinkmann MM, et al. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177(2):265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452(7184):234–238. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- 12.Itoh H, et al. UNC93B1 physically associates with human TLR8 and regulates TLR8-mediated signaling. PLoS ONE. 2011;6(12):e28500. doi: 10.1371/journal.pone.0028500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee BL, et al. UNC93B1 mediates differential trafficking of endosomal TLRs. eLife. 2013;2:e00291. doi: 10.7554/eLife.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7(1):49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 15.Mouchess ML, et al. Transmembrane mutations in Toll-like receptor 9 bypass the requirement for ectodomain proteolysis and induce fatal inflammation. Immunity. 2011;35(5):721–732. doi: 10.1016/j.immuni.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi F, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410(6832):1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 17.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167(4):1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 18.Feuillet V, et al. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc Natl Acad Sci USA. 2006;103(33):12487–12492. doi: 10.1073/pnas.0605200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uematsu S, et al. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat Immunol. 2006;7(8):868–874. doi: 10.1038/ni1362. [DOI] [PubMed] [Google Scholar]
- 20.Vijay-Kumar M, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117(12):3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uematsu S, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol. 2008;9(7):769–776. doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 22.Burdelya LG, et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science. 2008;320(5873):226–230. doi: 10.1126/science.1154986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shibata T, et al. PRAT4A-dependent expression of cell surface TLR5 on neutrophils, classical monocytes and dendritic cells. Int Immunol. 2012;24(10):613–623. doi: 10.1093/intimm/dxs068. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, et al. Acidic amino acid residues in the juxtamembrane region of the nucleotide-sensing TLRs are important for UNC93B1 binding and signaling. J Immunol. 2013;190(10):5287–5295. doi: 10.4049/jimmunol.1202767. [DOI] [PubMed] [Google Scholar]
- 25.Lee J, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8(12):1327–1336. doi: 10.1038/ncb1500. [DOI] [PubMed] [Google Scholar]
- 26.Lindau D, Rönnefarth V, Erbacher A, Rammensee HG, Decker P. Nucleosome-induced neutrophil activation occurs independently of TLR9 and endosomal acidification: Implications for systemic lupus erythematosus. Eur J Immunol. 2011;41(3):669–681. doi: 10.1002/eji.201040593. [DOI] [PubMed] [Google Scholar]
- 27.Onji M, et al. An essential role for the N-terminal fragment of Toll-like receptor 9 in DNA sensing. Nat Commun. 2013;4:1949. doi: 10.1038/ncomms2949. [DOI] [PubMed] [Google Scholar]
- 28.Pohar J, Pirher N, Benčina M, Manček-Keber M, Jerala R. The role of UNC93B1 protein in surface localization of TLR3 receptor and in cell priming to nucleic acid agonists. J Biol Chem. 2013;288(1):442–454. doi: 10.1074/jbc.M112.413922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Funami K, et al. The cytoplasmic ‘linker region’ in Toll-like receptor 3 controls receptor localization and signaling. Int Immunol. 2004;16(8):1143–1154. doi: 10.1093/intimm/dxh115. [DOI] [PubMed] [Google Scholar]
- 30.Fukui R, et al. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J Exp Med. 2009;206(6):1339–1350. doi: 10.1084/jem.20082316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Letran SE, et al. TLR5 functions as an endocytic receptor to enhance flagellin-specific adaptive immunity. Eur J Immunol. 2011;41(1):29–38. doi: 10.1002/eji.201040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Latz E, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5(2):190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 33.Hessa T, et al. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450(7172):1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.