

Significance
Kinesin regulation by autoinhibition has been extensively studied. However, it is known that cargo-bound kinesin motion can be altered by various signaling pathways. How are these kinesins regulated? Kinesin regulation by the signaling kinase casein kinase 2 (CK2) was previously reported to activate inactive kinesin at the level of the single motor head domain, but the mechanism was unknown. Here, using a multidisciplinary approach, we discover that kinesin inactivation involves a specific conformational change in the molecule’s neck linker, which controls microtubule affinity and is reversed by CK2.
Keywords: cargo travel, multiple-motor transport, transport regulation
Abstract
Kinesin is the canonical plus-end microtubule motor and has been the focus of intense study since its discovery in 1985. We previously demonstrated a time-dependent inactivation of kinesin in vitro that was fully reversible by the addition of purified casein kinase 2 (CK2) and showed that this inactivation/reactivation pathway was relevant in cells. Here we show that kinesin inactivation results from a conformational change that causes the neck linker to be positioned closer to the motor domain. Furthermore, we show that treatment of kinesin with CK2 prevents and reverses this repositioning. Finally, we demonstrate that CK2 treatment facilitates ADP dissociation from the motor, resulting in a nucleotide-free state that promotes microtubule binding. Thus, we propose that kinesin inactivation results from neck-linker repositioning and that CK2-mediated reactivation results from CK2’s dual ability to reverse this repositioning and to promote ADP release.
Intracellular microtubule-based transport is crucial for the creation and maintenance of cellular order, and altered transport is implicated in both neurodegeneration and cancer. Frequently, in vivo cargos are moved by multiple microtubule-based molecular motors (1–6), and changing the number of active motors on the cargo can change cargo force production (4) and also potentially the mean travel distance for predominantly unidirectionally moving cargos (7). However, until recently, it has been unclear how activity of cargo-bound motors might be regulated.
Transport is frequently regulated by signaling cascades [see, e.g., cAMP control of pigment granule transport (8) or APP transport (9)]. Thus, multiple signaling pathways might contribute to control of transport under different conditions, and signaling altered in disease might affect transport, which could then contribute to disease progression. Nonetheless, mechanistic understanding of such effects is limited. For these reasons, we would like to understand transport roles of specific disease-relevant kinases. One such kinase is casein kinase 2 (CK2), which is involved in development (10), is up-regulated in various cancers (11), and is decreased in neurodegeneration (12). We found that, over time, kinesin loses its ability to bind microtubules (becomes “inactive”) and that this loss of activity could be reversed by CK2 (13).
Mechanistically, how kinesin became inactive—and what CK2 did to reactivate it—was unknown. Here we discover that kinesin’s inactivation results from a conformational change involving repositioning of the neck linker (NL) and that reactivation reverses this conformational change. Intriguingly, the conformational change that results in reactivation causes release of ADP, converting kinesin from a weak microtubule-interacting state (K⋅ADP) to a strong one (K), so that in some ways CK2 acts like a small G-protein nucleotide-exchange factor.
Results
Kinesin-1 Inactivation Is Accelerated at Physiologic Temperatures.
Previously, we reported that when stored (“aged”) on ice, kinesin went inactive over the course of a few hours and lost the ability to bind to microtubules (13). New experiments have confirmed such inactivation and have found that it is accelerated at higher temperatures as measured by both microtubule pelleting assays of dimers (Fig. 1A) and microtubule (MT)-stimulated ATPase rates of monomers (Fig. 1 B and C).
Fig. 1.
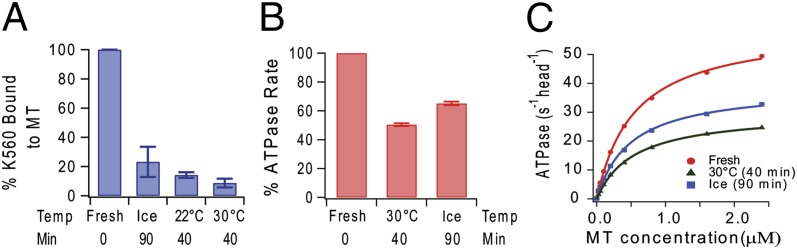
Kinesin inactivates more rapidly at higher temperatures. (A) Freshly thawed kinesin dimer is active (t = 0; left bar) and sediments with microtubules in the presence of AMP-PNP. Aging for 1.5 h (on ice) or 40 min (at warmer temperatures) resulted in decreased interactions with microtubules (all other bars), with increasing temperature resulting in accelerated inactivation. (B) The decrease in MT association detected by MT pelleting in A was accompanied by a decrease in MT-stimulated ATPase activity of K349 monomer. (C) Original data are summarized in B.
Inactive Kinesin Does Not Lose the Ability to Exchange Nucleotides.
Because ADP-bound kinesin has low MT affinity, one potential mechanism for kinesin inactivation involves kinesin becoming trapped in an ADP-bound state and losing its ability for nucleotide exchange. To test this hypothesis, we used α-P32–labeled ADP to directly measure the ability of active vs. inactive kinesin to release ADP. First, we incubated kinesin with α-P32–ATP, which is converted to α-P32–ADP. We took a sample of kinesin loaded with hot ADP and split it into two subsamples. One was immediately chased with excess cold ATP, and we found that kinesin could efficiently exchange nucleotides when the kinesin was in the active state at this early time point (Fig. 2A, left bars). The remaining sample was aged on ice for 4 h, to ensure sufficient inactivation, and was then chased with excess cold ATP. At this later time point, kinesin (that is mostly inactive) could still efficiently exchange hot ADP for cold (Fig. 2A, right bars). Thus, even when kinesin had lost its ability to bind to MT and was inactive, it could still exchange nucleotides. Note that these exchange experiments were performed in the absence of microtubules. Because the inactive kinesin loses the ability to interact with microtubules, the MT-stimulated ATPase activity does decrease over time (Fig. 1 B and C), so a difference in nucleotide exchange rate would be expected if microtubules were present.
Fig. 2.

Nucleotides do not play a central role in kinesin inactivation. (A) Using radiolabeled ATP in a pulse-chase dot-blot experiment, we tested kinesin’s ability to exchange nucleotides before and after aging on ice. Kinesin’s ability to exchange nucleotides was not impaired at later time points. (B and C) Kinesin inactivation was not dependent on the nucleotide state of the motor because kinesin purified away from bound nucleotides (via EDTA treatment) also loses its ability to bind MT over time on ice as measured by MT pelleting assay (B) and at approximately the same rate as normally purified kinesin (nucleotide bound) (C). Note that the pelleting assay in B (and K560–ADP assay in C) was performed without addition of AMP-PNP.
To further assess the importance of the nucleotide state in the inactivation process, we examined inactivation of kinesin without nucleotides. We purified nucleotide-free kinesin by incubating freshly purified kinesin with excess EDTA; EDTA was then removed by using a desalting column, glycerol was added, and the kinesin was flash frozen. At t = 0, we confirmed that it was both active and functionally nucleotide-free by carrying out a MT-pelleting experiment in the absence of adenosine 5′-(β, γ-imidio)triphosphate (AMP-PNP) (taking advantage of the fact that the nucleotide-free state is a high-MT-affinity state) and observing that almost all kinesin present was found in the MT pellet (Fig. 2B, left bar). Nonetheless, when aged, this kinesin became inactive (Fig. 2B), and its inactivation kinetics were similar to the inactivation of initially ADP-bound kinesin (Fig. 2C). Combined, these data suggest that some form of conformational change likely causes kinesin inactivation, rather than simply getting trapped in the ADP-bound state.
CK2 Accelerates Kinesin–ADP Exchange.
In our previous publication (13), we established that CK2 could both prevent inactivation, and reactivate already inactive kinesin. So what does CK2 do to reactivate the kinesin? One possible explanation is that CK2 accelerates the rate of nucleotide exchange; the pull-down experiments are too slow to test kinetic effects. To test this explanation, we examined kinesin’s basal ATPase rate. Without microtubules, kinesin’s ADP release is quite slow (on the order of minutes) and thus limits basal ATPase activity (14). If CK2 promotes ADP release, its presence might increase basal activity. We thus carried out a traditional ATPase assay, using fresh (active) kinesin. Our measurement of basal ATPase activity for kinesin alone was 0.011 s−1, consistent with past reports. Intriguingly, in the presence of CK2, the measured ATPase rate was 0.016 s−1, an increase of 35% relative to ATPase activity without CK2 (Fig. 3A). This result cannot be explained by the CK2 ATPase activity (because that was minimal—0.003 s−1) or by CK2’s phosphorylation of kinesin, because the same 35% increase in kinesin’s ATPase activity was observed even in the presence of tetrabromocinnamic acid (TBCA), a drug that inhibits CK2 enzymatic activity (13, 15). Thus, in the absence of microtubules, CK2 appears to accelerate kinesin’s ADP release.
Fig. 3.

CK2 accelerates kinesin nucleotide exchange, but nucleotide exchange is not required for reactivation. (A) Kinesin treated with CK2 shows an increase in basal ATPase rate as measured by a regenerating ATP system. Importantly, this increase was observed in the presence of the CK2-specific ATP competitive inhibitor TBCA, indicating that CK2 kinase activity is not responsible for the observed increase in kinesin ATPase activity. All ATPase measurements were collected with freshly thawed kinesin. *P < 0.05; **P < 0.005. (B) To directly visualize nucleotide exchange, kinesin loaded with radiolabeled nucleotide was treated with CK2α and showed loss of radiolabeled nucleotide compared with untreated samples after aging on ice. (C) In addition to native-PAGE, dot-blot pulse-chase experiments found that kinesin treated with CK2 holoenzyme showed a significant reduction in radiolabeled kinesin after aging on ice. (D) Quantified signal from C. (E) Nucleotide-free kinesin aged for 4 h on ice (as per Fig. 2 B and C) can also be reactivated with treatment of CK2 added halfway through aging, as measured by MT pelleting assays. (F) TBCA treatment of kinesin also did not affect the ability of CK2 to reactivate kinesin. Combined, the data show that CK2-mediated reactivation of kinesin is not dependent on its ability to facilitate nucleotide exchange.
To confirm directly that CK2 facilitates faster nucleotide exchange, we returned to radioactive ATP. We loaded freshly thawed kinesin with α-P32–ATP for 15 min at room temperature, and kinesin was then split into two samples. One was treated with CK2α and the other was left untreated. The proteins were aged on ice for 2 h and then run on a native gel, in order to keep intact the nucleotide pocket (keeping in place the radiolabeled nucleotide, if it was present). Signal was then detected by using a PhosphorImager cassette and Typhoon scanner. Kinesin untreated with CK2 showed a significant amount of radiolabeled protein. However, kinesin treated with CK2 showed a significant reduction in radiolabeled signal (Fig. 3B). CK2α was used rather than holoenzyme, because in contrast to kinesin–CK2, the kinesin–CK2α interaction is transient (13), so CK2α and kinesin run separately in the native gels.
To eliminate the possibility that CK2 was simply hydrolyzing the ATP so that there was simply less ATP available to bind the kinesin in the presence of CK2, we repeated the experiment using a dot-blot method in a pulse-chase format. Briefly, kinesin was loaded as before. The protein was then split into two samples—treated and untreated with CK2. To each, excess cold ATP was added. After 2 h aging on ice, the samples were dotted onto nitrocellulose, excess sample was vacuumed through, and dots were thoroughly washed with buffer. The nitrocellulose was again imaged on a PhosphorImager cassette. Visualization of the nitrocellulose found that samples treated with CK2 had a highly statistically significant reduction in protein that was radiolabeled (Fig. 3 C and D). Both the native gel and dot-blot assay thus directly visualized changes in hot-nucleotide-bound protein, indicating that CK2 promotes release of nucleotide from kinesin in a microtubule-independent manner.
Is this CK2-induced nucleotide exchange important for kinesin reactivation? To address this question, we tested whether the CK2 was able to reactivate the inactive nucleotide-free kinesin aged 4 h on ice (Fig. 2B). Indeed, CK2 treatment reactivated the nucleotide-free kinesin, which had no nucleotide to release (Fig. 3E). Further, CK2 was able to reactivate kinesin even in the presence of TBCA, a CK2-specific kinase inhibitor that functions as an ATP competitor (13) (Fig. 3F). Combined, these data show that, although nucleotide exchange is affected by the presence of CK2, it is not required for motor reactivation. Instead, we hypothesize that CK2 binding results in a conformational change of the motor and that it is this change that activates the kinesin, in addition to resulting in nucleotide exchange.
CK2 Facilitates Kinesin–MT Association but Not via Direct Recruitment.
Normally, kinesin in solution is bound to ADP and interacts weakly with microtubules: Without excess ATP, even when it is potentially fully active, only ∼20% of the protein precipitates with microtubules in pull-down experiments (Fig. 4A, left bar). Our data above suggest that because CK2 promotes ADP release, it might increase the affinity of kinesin for MTs by increasing the percentage of kinesin in solution in the nucleotide-free state (which would then have a high MT affinity). To test this hypothesis, we took normally purified kinesin (unaged, that was all presumably active) and split the sample into multiple aliquots. To the first, we mock-CK2–treated it, aging it for 2 h on ice but adding nothing, and then carried out a microtubule coprecipitation assay. Because the kinesin had been purified normally, it presumably was bound to ADP (14), and ∼20% of the total kinesin coprecipitated (Fig. 4A, first bar). Under the same conditions, but with AMP-PNP before the pull-down, we saw an increase in the amount of kinesin coprecipitating on MT (Fig. 4A, second bar); the overall amount precipitating was reduced relative to the freshly thawed kinesin (Fig. 4A) due to inactivation that occurred during the experiment. Finally, an aliquot was treated with CK2 and then subjected to identical coprecipitation, but, importantly, with no added AMP-PNP. Even without AMP-PNP, the majority of the kinesin coprecipitated with microtubules (Fig. 4A, right bar), consistent with the above data that CK2 promoted ADP release, increasing the motor’s affinity for the microtubules.
Fig. 4.

CK2 reactivation of kinesin is independent of a direct recruitment via MT-bound CK2. (A) Freshly thawed kinesin (t = 0) requires the presence of the stabilizing nucleotide AMP-PNP to bind MT. Over time, kinesin aged on ice loses its ability to bind MT, even in the presence of the AMP-PNP (center bars). CK2 treatment decouples MT sedimentation from the required presence of AMP-PNP (right bars), consistent with the hypothesis that CK2 facilitates nucleotide release, as measured by kinesin’s ability to bind MT in the absence of AMP-PNP. (B) To test whether CK2 linked kinesin to MT directly, we titrated in increasing amounts of ADP to alter kinesin binding if the motor was binding with its own MT affinity. No AMP-PNP was present. (C) Western blot showing that increasing ADP did not change the amount of CK2 bound to MT, so changes in the MT-bound kinesin (B) were due to the ADP changing kinesin to the low-MT-affinity state. ANOVA confirmed that the differences were significant.
Because CK2 is able to bind both microtubules and motors, in principle the increased fraction of motors in the pellet could reflect direct recruitment to the MT via CK2, rather than actual alteration of motor activity. To test this possibility, we examined the effect of CK2 in the presence of increasing amounts of ADP. The coprecipitation experiments clearly showed that ADP addition blocked the CK2-mediated increase in kinesin’s coprecipitation, in a dose-dependent fashion (Fig. 4B). This experiment had two interpretations: Either kinesin was binding directly to the microtubules so that its nucleotide state mattered, or, alternatively, it was being linked to the microtubules via CK2, but ADP was displacing CK2 from the microtubule. We thus tested whether ADP affected the amount of MT-bound CK2. The coprecipitation experiment of CK2 and MT showed that it did not (Fig. 4C). Because CK2 is still on the MT, but the increased ADP drives the kinesin off the microtubule, we conclude that the experiments are consistent with the hypothesis that the increase in MT-bound kinesin is due to CK2’s effect on kinesin’s nucleotide state, rather than due to a direct linkage of kinesin to the microtubule via CK2.
Inactive Kinesin Undergoes a Conformational Change Resulting in Docking of the NL.
The above data indicate that it is unlikely that kinesin has become inactive due to becoming “locked” in a particular nucleotide state, but that CK2 does cause ADP release. One rationalization of these two observations is that, although kinesin inactivation/reactivation does not depend directly on kinesin’s nucleotide state, CK2 causes a conformational change that both activates the motor and causes ADP release. We previously ruled out gross conformational changes: Inactivation occurs at the level of the single head, and no large change was observed in the sedimentation characteristics of the protein (13). Instead, we hypothesize that a smaller change occurs. Recent work highlighted the position of the NL domain relative to the rest of the head as changing during the enzymatic cycle and as being important for alteration of the MT binding affinity (16, 17). Thus, kinesin inactivation might reflect a conformational change that alters the position/conformation of the NL relative to the head. To test this possibility, we carried out single-molecule FRET experiments. Previous work (18) established that fluorescently labeling residues, 43 (at the minus-end oriented tip of the catalytic core), 215 [at the plus-end oriented tip of the catalytic core of the motor domain (MD)], and 342 (just at the end of the NL) allowed accurate monitoring of the NL position relative to the MD. If kinesin inactivation involves changes in the conformation of the NL, use of the 215/342 and 43/342 FRET pairs should detect it.
The Cy3/Cy5 FRET signal was monitored from single MDs attached specifically to coverslips via antibody against C-terminal His-tag in the absence of ATP and microtubule. As controls, FRET-labeled kinesins were flowed onto axoneme MT fixed to coverslips, and the NL was docked by using AMP-PNP (Fig. 5 A and B). Using these samples as confirmed docking signals, we looked at the position of the kinesin NL relative to the motor head, with and without CK2 treatment.
Fig. 5.

Conformational change controlling NL position is altered by CK2. The relative position of the NL and MD can be monitored by Cy3–Cy5 single dye-pair FRET. Using 43/342 and 215/342 cysteine pairs introduced to monomeric Cys-light kinesin (see cartoon in A and B), we can monitor movement of the NL relative to the MD. (A and B) Binding labeled freshly thawed kinesin to axoneme MT in the presence of AMP-PNP served as a control to show docking of the NL. In all histograms, red represents the Gaussian fit for the docked signal, and blue is the fit for the undocked state. (C) Movement of the NL visualized using kinesin labeled at 215/342 cysteines attached to coverslip in the absence of microtubule. Freshly thawed kinesin began in the undocked position. (C′) After aging at 30 °C, the FRET efficiency distribution changed to one that had a significant contribution from a second peak, localized at ∼0.8 FRET efficiency (compare with docked signal in A). (C″) This docked-like conformation was prevented with CK2 treatment. (D′) Not only did CK2 prevent the docking of the NL, CK2 treatment could reverse docking of the NL. CK2 aged an additional 90 min on ice showed rescue of the docked NL. (D″) Kinesin left untreated had a different distribution of peak FRET efficiencies, with a new peak appearing at EFRET ∼0.6. (E) FRET data were validated with fluorescently labeled dimeric single kinesins moving on axoneme MT. Landing and movement events were counted in CK2-preincubated treated and untreated kinesin, as well as CK2-reactivated treated and untreated kinesin. (F) Kinesin labeled at 43/342 cysteines, freshly thawed, has an undocked EFRET peak at ∼0.8 (compare with docked state in B). (F′) Aged 43/342 kinesin shifted into the docked-like peak and was reversed with treatment by CK2 (F″).
For the 215/342 labeled kinesin examined immediately after thawing, we observed a distribution with relatively low EFRET (∼0.4) (Fig. 5C), but this distribution changed over time, so that after 40 min of aging at 30 °C, we observed a distribution with higher EFRET (Fig. 5C′). Importantly, the presence of CK2 prevented the shift (Fig. 5C″).
The 215/342 distributions (Fig. 5 C–C″) appeared to be composed of two Gaussians, potentially reflecting two states. To test this hypothesis, we used a nonlinear least-squares approach to fit each curve to the sum of two Gaussians; the exact amplitudes and peak locations were determined by fitting. In each case, the overall distribution was indeed well described by the sum of two Gaussians. The locations of the peaks were approximately the same, but the relative amplitude associated with each peak was different: For the FRET distributions corresponding to the immediately thawed and CK2-treated kinesin, the low-efficiency (∼0.4) FRET peak had high amplitude, but the high-efficiency (∼0.8) FRET peak had low amplitude. The relative amplitude of these two peaks were reversed for the kinesin aged without CK2. We thus hypothesize that the high-efficiency FRET signal—which was initially predominantly absent—corresponds to the inactive kinesin state. From this decomposition fitting, we can therefore use the area under each curve to estimate the percentage of the active vs. inactive kinesin. Immediately after thawing, the area under the 0.4 curve contains 85% of the total area, suggesting that almost all of the kinesin is active. In contrast, after aging, this 0.4 curve contained only 45% of the total population, so from this analysis, we expect ∼55% of the motors to be inactive. The 52% decrease in the low-FRET population after aging is consistent with the 50% decrease in the ATPase after 40 min of aging at 30 °C (Fig. 1B), demonstrating that the low FRET signal represents the active state. Finally, after CK2 treatment, again the majority of the kinesin was in the 0.4 curve, and we estimate that only 22% of the population is inactive.
In addition to preventing inactivation—where kinesin was aged with or without CK2—we tested the ability of CK2 to reactivate motors. Cy3/Cy5-labeled motors were first inactivated (Fig. 5C′) and then treated with CK2 for 90 min on ice. As expected, CK2-treated motors showed a decreased high FRET signal and a shift of FRET efficiency to ∼0.4 (Fig. 5D′). Interestingly, in the untreated motors (Fig. 5D″; which were aged significantly longer than the motors in Fig. 5C′), we saw the appearance of a third peak at an EFRET ∼0.6. More rigorous investigation will be required to fully determine what this FRET state represents, but it appears to reflect some longer-term evolution of the kinesin’s structure, because in multiple experiments we did not see it for the shorter aging periods.
These estimates of active vs. inactive motors can be compared with actual single-molecule motile activities by examining motility of single Cy3-labeled K490 Cys-light kinesins moving along axoneme MTs (Fig. 5E). Under identical measurement conditions, immediately after thawing, we observed a landing rate of 0.037 ± 0.003 fluorescent spots per second per micrometer of MT. With aging, this rate decreased to 0.017 ± 0.003 and with CK2 was restored to 0.027 ± 0.003. From the area under the FRET curves (in Fig. 5 C–C″), assuming that the 0.037 landing rate reflects activity of 85% of the active motors, for 45% active motors, we would then predict an observed landing rate of 0.020, and for 78% active motors we would predict 0.034. Comparing predictions to experimental results, we find that for the inactive states the two are within 15% of each other, and for the CK2-treated states they are within 25%. Although not perfect, given the noisy FRET data and the fact that the FRET analysis was carried out on single-headed constructs but the motility on dimeric motors, such agreement is encouraging.
Motion of the NL relative to the MD was monitored with the second set of FRET pairs 43/342 (minus-end of the catalytic core and the end of the NL). Freshly thawed 43/342 kinesin had a high FRET efficiency, consistent with being in the undocked position (Fig. 5F), but over time the NL shifted away from the minus end, toward the plus end (Fig. 5F′). Treatment by CK2 reversed the position of the NL, returning the NL to the undocked, high-efficiency FRET state (Fig. 5F″). Like the 215/342 FRET pair, the 43/342 pair showed similar distributions (compare Fig. 5 C–C″ to Fig. 5 F–F″).
The motion of the NL as monitored by the 43/342 FRET pair is thus consistent with observations made with the 215/342: Without CK2 treatment, the kinesin NL undergoes a conformational change, resulting in movement toward the MD, and adopting a conformation that may be the docked state, taken by the motor protein when active and bound to MT (compare Fig. 5A with Fig. 5C′ and Fig. 5B with Fig. 5F′). Note that because microtubules were absent in these studies, these FRET data indicate that CK2-mediated regulation of kinesin is not through complex formation on the MT.
Discussion
Mechanistic understanding of kinesin processivity and MT binding are relatively advanced, but regulation of kinesin activity is less well characterized. We previously described a novel phenomenon of a time-dependent inactivation of kinesin that was fully reversible by the addition of CK2 (13). Moreover, we demonstrated that CK2-mediated reactivation of kinesin was independent of CK2’s kinase activity and occurred at a 1:1 ratio of kinesin to CK2. This inactivation/reactivation involved only the monomeric kinesin heads. However, we did not know the molecular mechanism of kinesin inactivation/reactivation. Here, we find that kinesin inactivation results in a conformational change, where the NL is positioned close to the MD, and treatment of kinesin with CK2 was prevented/reversed this close approach between NL and MD. In addition, CK2 treatment facilitated nucleotide loss in the motor, favoring a nucleotide-free state that promoted MT binding. However, nucleotide exchange was not required for reactivating the motor, as demonstrated by reactivation of the nucleotide-free motor.
How should this conformational change be interpreted? For normally functioning dimeric kinesin on microtubule, binding of ATP (or AMP-PNP) by the MD normally results in docking of the NL to the MD; such a docked state is typically thought of as a strong MT interaction state. However, there are likely other conformations of the NL that position it close to the MD but that are different from this docked state that has been visualized by ATP-like crystal structures. One such conformation was previously induced by selective cross-linking, which artificially induced NL immobilization (via designed disulfide bonds) and led to the complete inhibition of kinetic activity for monomeric as well as dimeric kinesin constructs (17). Our inactivation—with concurrent loss of microtubule binding—could thus be similar to the decreased microtubule affinity observed previously when the NL was artificially held in place close to the MD (17). Thus, one interpretation of our data is that the NL is not truly “docked” but, rather, is adsorbed onto the MD in a different “off-pathway” conformation, reminiscent of the positioning of the Eg5 NL in the ADP-bound state (19).
However, we favor a second model, in which the close positioning of the NL and MD we observed does reflect correct (normal) docking. In this model, the aberrant function results from being in an unusual mixed conformation. There is accumulating work suggesting that it is possible to partially decouple ADP binding, NL docking to the MD, and conformational changes that occur at the motor’s microtubule binding surface: Although binding of ATP usually induces both NL docking and increased MT affinity, there are multiple crystal structures (20, 21) that are characterized as ATP-like and display a docked NL conformation but have ADP present in the nucleotide pocket. Further, these structures may not be compatible with microtubule binding (i.e., are weak affinity), because the recently solved true AMP-PNP–bound kinesin-1 structure [using a kinesin–tubulin complex crystal (22)] has a clearly different structure in the microtubule interface (especially α4 helix and loop 11, which is important for microtubule binding). This finding leads us to suggest that our inactive kinesin is an off-pathway “mixed” conformational state, bound to ADP but with a docked NL and low-affinity microtubule interface. In such a model, when in solution, ADP-bound kinesin is in equilibrium between the NL undocked (on-pathway) and docked (off-pathway) states, with a slow interconversion rate. Then, if it transits to the ADP-bound docked state after a long incubation period, the motor cannot bind to the microtubule, so that ADP release is prohibited. Consistent with such a model, Sindelar et al. (21) showed that ∼20% of ADP-bound kinesin in solution is in the NL docked conformation, even in the absence of microtubules. CK2 then prevents this interconversion or, alternatively, reactivates the inactive docked state, possibly by binding to and unzipping the NL or by binding near the nucleotide pocket to induce ADP release. Preferential binding of CK2 to the disordered NL may then allosterically induce ADP release from the nucleotide pocket, as several studies have already suggested (23–26), resulting in a nucleotide-free undocked state followed by immediate ATP binding and hydrolysis to return to the on-pathway ADP-bound undocked state.
It will be exciting to investigate detailed differences between the transient NL docking occurring during normal kinesin activity and the more stable NL docking associated with our inactive state, and ultimately to determine which of the above models is correct.
Why does kinesin go inactive? The inactive state does appear more thermodynamically stable, and at first such inactivation appears to be an inefficient strategy for a motor. Although work remains, we hypothesize that such gradual inactivation allows for additional cellular control of when and where kinesin is active. Rather than being uniformly spread throughout the cell, CK2 is found in specific subcellular compartments (27). In at least one instance, this localization is controlled via a combination of phosphorylation and selective interactions with a recruitment protein (28). In principle, then, such localization allows for local up-regulation of kinesin function at specific subcellular locations, making possible spatial transport control (via CK2 localization) as well as temporal transport control (via overall CK2 levels). The extent to which cells take advantage of such a scheme to direct cargo motion will be an exciting new area of study. It is also possible that the disordered (undocked) NL of ADP-bound kinesin is unfavorable in the crowded environment inside the cell, because it is likely to cause nonspecific interactions and aggregation. In principle, then, the transition to the inactive/docked state may serve to prevent such nonspecific interactions. CK2 binding may be one such—functionally significant—interaction, which also prevents futile ATP hydrolysis while kinesin is in an idling state.
Methods
Protein Purification.
Functional, tailless kinesin was expressed in terrific broth (29) and purified as described (13, 30). Nucleotide-free kinesin was made by incubating normally purified kinesin with excess EDTA. EDTA was removed by buffer exchange (GE; PD10).
Bovine tubulin was purified over a phosphocellulose column as described (31). Recombinant CK2 was purchased from New England Biolabs.
CK2 Preincubation and Reactivation.
CK2 preincubation experiments were performed as described (13). Briefly, kinesin was incubated with CK2, on ice or at higher temperatures, by using untreated samples as controls. Motor activity was determined by MT affinity pull-down and quantitative westerns. Antibodies used were AKIN01 (Cytoskeleton), CK2α (Cell Signaling), and AA43.1 (Hybridoma Bank). TBCA (EMD) was added to samples during preincubation. For reactivation, kinesin was first inactivated by incubation alone and then treated with CK2.
ATPase Assay.
For basal kinesin ATPase rates, an ATP-regenerating system was used (32).
Microtubule Affinity Pull-Down.
Kinesin activity was measured by its ability to bind to and pellet with MT. Assays were performed as described (13, 33).
Radiolabeled Kinesin.
Radiolabeled kinesin experiments were modified from Gilbert and Johnson (34). Briefly, purified K560 was incubated with α-P32–ATP (Perkin-Elmer) at room temperature, to load up K560 with radiolabeled nucleotide. Kinesin was then treated with CK2α for 90 min on ice, and untreated samples were used as controls. Samples were then run on a native gel (Bio-Rad), and signal intensities were quantified by using ImageQuant. For dot-blot pulse-chase experiments, kinesin was loaded as before. Kinesin was then chased with excess cold ATP at different time points. Samples were dotted onto blocked nitrocellulose. Excess sample was pulled through by vacuum and washed with buffer (20 mM Hepes, pH 7.2, 5 mM magnesium acetate, 0.1 mM EDTA, 0.1 mM EGTA, 50 mM potassium acetate, 1 mM DTT). Nitrocellulose was then imaged, and signal was quantified by using ImageQuant. For CK2-treated samples, K560 was loaded as and chased as before, with or without CK2. Samples were imaged on the Typhoon scanner and quantified with ImageQuant (GE).
Single-Molecule FRET.
Single-molecule FRET observation and analysis were carried out by using the Cys-light kinesin monomer (349-residue with C-terminal His6 tag) with cysteines introduced into 215/342 or 43/342 residues as described (18), except that fluorescently labeled kinesins were immobilized on the glass surface via anti-His antibodies in the absence of microtubule. Flow cell constructed between a quartz slide and a coverslip was first filled with 25 μg/mL 6× His monoclonal antibody (010-21861; Wako) in BRB12 buffer [12 mM Pipes–KOH (pH 6.8), 2 mM MgCl2, 1 mM EGTA] for 3 min, followed by a washing with 1 mg/mL casein in BRB12 for 3 min. Then, 150 nM Cy3/Cy5-labeled kinesin after incubation in CK2 reaction buffer [20 mM Tris⋅HCl (pH 7.5), 50 mM KCl, 10 mM MgCl2, 0.5 mM EGTA] was diluted to 100 pM with BRB12 buffer and flowed into the chamber for 3 min. After washing, the chamber was filled with imaging buffer (BRB12 supplemented with 4.5 mg/mL glucose, 50 U/mL glucose oxidase, 50 U/mL catalase, 0.5% β-mercaptoethanol, and 1 mM ATP) and sealed. FRET data were collected at 22 °C.
Supplementary Material
Acknowledgments
We thank Herb Miller and Dr. Les Wilson for their continued support. S.P.G. thanks Dr. Steven Rosenfeld (Cleveland Clinic) for helpful conversations. This work was supported by National Institutes of Health Grants R01-GM64624 (to S.P.G.) and P50-GM76516 (to S.P.G. and L.B.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
References
- 1.Miller RH, Lasek RJ. Cross-bridges mediate anterograde and retrograde vesicle transport along microtubules in squid axoplasm. J Cell Biol. 1985;101(6):2181–2193. doi: 10.1083/jcb.101.6.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashkin A, Schütze K, Dziedzic JM, Euteneuer U, Schliwa M. Force generation of organelle transport measured in vivo by an infrared laser trap. Nature. 1990;348(6299):346–348. doi: 10.1038/348346a0. [DOI] [PubMed] [Google Scholar]
- 3.Kural C, et al. Kinesin and dynein move a peroxisome in vivo: A tug-of-war or coordinated movement? Science. 2005;308(5727):1469–1472. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- 4.Shubeita GT, et al. Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell. 2008;135(6):1098–1107. doi: 10.1016/j.cell.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hendricks AG, et al. Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr Biol. 2010;20(8):697–702. doi: 10.1016/j.cub.2010.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ori-McKenney KM, Xu J, Gross SP, Vallee RB. A cytoplasmic dynein tail mutation impairs motor processivity. Nat Cell Biol. 2010;12(12):1228–1234. doi: 10.1038/ncb2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bullock SL, Nicol A, Gross SP, Zicha D. Guidance of bidirectional motor complexes by mRNA cargoes through control of dynein number and activity. Curr Biol. 2006;16(14):1447–1452. doi: 10.1016/j.cub.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 8.Rodionov V, Yi J, Kashina A, Oladipo A, Gross SP. Switching between microtubule- and actin-based transport systems in melanophores is controlled by cAMP levels. Curr Biol. 2003;13(21):1837–1847. doi: 10.1016/j.cub.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 9.Fu MM, Holzbaur ELF. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J Cell Biol. 2013;202(3):495–508. doi: 10.1083/jcb.201302078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerra B, Issinger O-G. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis. 1999;20(2):391–408. doi: 10.1002/(SICI)1522-2683(19990201)20:2<391::AID-ELPS391>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 11.Wang G, Unger G, Ahmad KA, Slaton JW, Ahmed K. Downregulation of CK2 induces apoptosis in cancer cells—a potential approach to cancer therapy. Mol Cell Biochem. 2005;274(1-2):77–84. doi: 10.1007/s11010-005-3077-1. [DOI] [PubMed] [Google Scholar]
- 12.Blanquet PR. Casein kinase 2 as a potentially important enzyme in the nervous system. Prog Neurobiol. 2000;60(3):211–246. doi: 10.1016/s0301-0082(99)00026-x. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, et al. Casein kinase 2 reverses tail-independent inactivation of kinesin-1. Nat Commun. 2012;3:754. doi: 10.1038/ncomms1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hackney DD. Kinesin ATPase: Rate-limiting ADP release. Proc Natl Acad Sci USA. 1988;85(17):6314–6318. doi: 10.1073/pnas.85.17.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pagano MA, et al. Tetrabromocinnamic acid (TBCA) and related compounds represent a new class of specific protein kinase CK2 inhibitors. ChemBioChem. 2007;8(1):129–139. doi: 10.1002/cbic.200600293. [DOI] [PubMed] [Google Scholar]
- 16.Tomishige M, Vale RD. Controlling kinesin by reversible disulfide cross-linking. Identifying the motility-producing conformational change. J Cell Biol. 2000;151(5):1081–1092. doi: 10.1083/jcb.151.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hahlen K, et al. Feedback of the kinesin-1 neck-linker position on the catalytic site. J Biol Chem. 2006;281(27):18868–18877. doi: 10.1074/jbc.M508019200. [DOI] [PubMed] [Google Scholar]
- 18.Tomishige M, Stuurman N, Vale RD. Single-molecule observations of neck linker conformational changes in the kinesin motor protein. Nat Struct Mol Biol. 2006;13(10):887–894. doi: 10.1038/nsmb1151. [DOI] [PubMed] [Google Scholar]
- 19.Turner J, et al. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J Biol Chem. 2001;276(27):25496–25502. doi: 10.1074/jbc.M100395200. [DOI] [PubMed] [Google Scholar]
- 20.Kozielski F, et al. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91(7):985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 21.Sindelar CV, et al. Two conformations in the human kinesin power stroke defined by X-ray crystallography and EPR spectroscopy. Nat Struct Biol. 2002;9(11):844–848. doi: 10.1038/nsb852. [DOI] [PubMed] [Google Scholar]
- 22.Gigant B, et al. Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat Struct Mol Biol. 2013;20(8):1001–1007. doi: 10.1038/nsmb.2624. [DOI] [PubMed] [Google Scholar]
- 23.Hyeon C, Onuchic JN. Mechanical control of the directional stepping dynamics of the kinesin motor. Proc Natl Acad Sci USA. 2007;104(44):17382–17387. doi: 10.1073/pnas.0708828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyeon C, Onuchic JN. Internal strain regulates the nucleotide binding site of the kinesin leading head. Proc Natl Acad Sci USA. 2007;104(7):2175–2180. doi: 10.1073/pnas.0610939104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uemura S, Ishiwata S. Loading direction regulates the affinity of ADP for kinesin. Nat Struct Biol. 2003;10(4):308–311. doi: 10.1038/nsb911. [DOI] [PubMed] [Google Scholar]
- 26.Jana B, Hyeon C, Onuchic JN. The origin of minus-end directionality and mechanochemistry of Ncd motors. PLOS Comput Biol. 2012;8(11):e1002783. doi: 10.1371/journal.pcbi.1002783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faust M, Montenarh M. Subcellular localization of protein kinase CK2. A key to its function? Cell Tissue Res. 2000;301(3):329–340. doi: 10.1007/s004410000256. [DOI] [PubMed] [Google Scholar]
- 28.St-Denis NA, Bailey ML, Parker EL, Vilk G, Litchfield DW. Localization of phosphorylated CK2alpha to the mitotic spindle requires the peptidyl-prolyl isomerase Pin1. J Cell Sci. 2011;124(Pt 14):2341–2348. doi: 10.1242/jcs.077446. [DOI] [PubMed] [Google Scholar]
- 29.Brodsky O, Cronin CN. Economical parallel protein expression screening and scale-up in Escherichia coli. J Struct Funct Genomics. 2006;7(2):101–108. doi: 10.1007/s10969-006-9013-0. [DOI] [PubMed] [Google Scholar]
- 30.Case RB, Pierce DW, Hom-Booher N, Hart CL, Vale RD. The directional preference of kinesin motors is specified by an element outside of the motor catalytic domain. Cell. 1997;90(5):959–966. doi: 10.1016/s0092-8674(00)80360-8. [DOI] [PubMed] [Google Scholar]
- 31.Miller HP, Wilson L. Preparation of microtubule protein and purified tubulin from bovine brain by cycles of assembly and disassembly and phosphocellulose chromatography. Methods Cell Biol. 2010;95:3–15. doi: 10.1016/S0091-679X(10)95001-2. [DOI] [PubMed] [Google Scholar]
- 32.Hackney DD, Jiang W. Assays for kinesin microtubule-stimulated ATPase activity. Methods Mol Biol. 2001;164:65–71. [PubMed] [Google Scholar]
- 33.Huang TG, Hackney DD. Drosophila kinesin minimal motor domain expressed in Escherichia coli. Purification and kinetic characterization. J Biol Chem. 1994;269(23):16493–16501. [PubMed] [Google Scholar]
- 34.Gilbert SP, Johnson KA. Expression, purification, and characterization of the Drosophila kinesin motor domain produced in Escherichia coli. Biochemistry. 1993;32(17):4677–4684. doi: 10.1021/bi00068a028. [DOI] [PubMed] [Google Scholar]