

Abstract
Background/Aims
It is increasingly recognized that there is sexual dimorphism in kidney disease progression; however, this disparity is lost in the presence of diabetes where women progress at a similar rate to men. The renin-angiotensin-aldosterone system (RAAS) is known to regulate diabetes-induced kidney injury, and recent literature would suggest that gender differences exist in RAAS-dependent responses in the kidney. In this regard, these gender differences may be overcome by excessive salt intake. Thereby, we hypothesized that salt would promote proteinuria in transgenic female rats under conditions of excess tissue angiotensin (Ang) II and circulating aldosterone.
Materials and Methods
We utilized young female transgenic (mRen2)27 (Ren2) rats and Sprague-Dawley (SD) littermates and fed a high-salt diet (4%) over 3 weeks.
Results
Compared to SD and Ren2 controls, female Ren2 rats fed a high-salt diet displayed increases in proteinuria, periarterial and interstitial fibrosis as well as ultrastructural evidence of basement membrane thickening, loss of mitochondrial elongation, mitochondrial fragmentation and attenuation of basilar canalicular infoldings. These findings occurred temporally with increases in transforming growth factor-β but not indices of oxidant stress.
Conclusions
Our current data suggest that a diet high in salt promotes progressive kidney injury as measured by proteinuria and fibrosis associated with transforming growth factor-β under conditions of excess tissue Ang II and circulating aldosterone.
Key Words : Angiotensin II, Transgenic (mRen2)27 rat, Proteinuria, Fibrosis, Reactive oxygen species
Introduction
There is increasing interest in sexual dimorphism as it impacts cardiovascular and kidney disease progression. In this context, previous work would suggest that men are more susceptible to kidney disease progression and increases in proteinuria than women [1,2]. This is true for numerous disease states such as membranous, polycystic kidney, or even nondiabetic kidney disease; however, it is not as clear for diabetic kidney disease where the rate of progression is similar between males and females [2]. In this context, little is known as to why women have a propensity to progress and develop proteinuria at a rate similar to men in the presence of diabetes.
There is sufficient data from human and experimental models to support the concept that progression of diabetic kidney disease, as measured by proteinuria, is tightly regulated by the renin-angiotensin-aldosterone system (RAAS) [3,4]. In both clinical and preclinical models, interruption of the RAAS has been shown to reduce proteinuria and slow progression of diabetic kidney disease supporting a distinct role for the RAAS [3,4,5]. In this regard, both angiotensin (Ang) II and aldosterone induce progressive kidney injury through increases in proteinuria and are morphologically characterized by interstitial fibrosis through activation of transforming growth factor (TGF)-β [6,7,8]. While TGF-β has been implicated in interstitial fibrosis, there has been work to suggest that TGF-β induces epithelial-mesenchymal transition (EMT) as a contributing mechanism [9,10].
In addition, data from experimental models suggest that there are gender differences in how the kidney responds to components of the RAAS, wherein estrogen has been shown to attenuate the impact of Ang II on proximal tubule-dependent Ang type 1 receptor responses [11]. To this end, others and we have previously observed in a model of RAAS overactivation that there exists dimorphism in blood pressure and proteinuria responses between males and females [12,13]. In this regard, the differences observed in proteinuria between female and male congenic mRen(2)27 (Ren2): Lewis rats were overcome by administering excess salt. Further, in combination with aldosterone, salt potentiates Rho kinase-dependent activation of TGF-β and downstream fibrosis [8].
In addition to intake of excess fat and carbohydrate, salt in excess contributes much to our Western diet and the high incident rate of type 2 diabetes and chronic kidney disease. The kidney serves to regulate excess salt and there have been decades of literature discussing the importance of salt restriction and blood pressure control. However, there is very little information on salt intake and kidney injury and even less on gender effects of salt intake [14]. In order to address the effect of increased dietary salt intake on proteinuria and interstitial fibrosis in female rats, we utilized the transgenic Ren2 rat that overexpresses the mouse renin transgene and manifests elevated plasma aldosterone levels as well as increased kidney tissue Ang II. The female Ren2 rat exhibits normal body mass, but develops insulin resistance and hypertension, yet little proteinuria [12,15,16]. Therefore, to investigate the impact of increased dietary salt on the female kidney structure and function, we fed female Ren2 rats with a high-salt diet over 3 weeks and examined proteinuria along with structural abnormalities associated with progressive kidney disease.
Materials and Methods
Animals and Treatments
All animal procedures were approved by the animal care and use committees at the University of Missouri and the Harry S. Truman Memorial Veterans' Hospital and housed in accordance with NIH guidelines. Female Ren2 (6-7 weeks of age) and age-matched female Sprague-Dawley (SD) rats were randomly assigned to feed on normal chow (Ren2-C and SD-C, respectively) or chow containing 4% of salt (Ren2-HS and SD-HS, respectively) for 21 days [16].
Urine Measures
Both creatinine and protein concentrations in urine were analyzed on an automated clinical chemistry analyzer (AU680; Olympus America Inc., Centerville, Pa., USA) using commercial assays [17]. The chemistry instrument was calibrated and proper controls were performed before analysis.
Quantification of Periarterial and Interstitial Fibrosis
A total of 5 μm sections of kidney from all treatments were stained with picrosirius red, according to the manufacturer's procedure. Slides were checked with a Nikon50i microscope, and 10 images were randomly captured from proximal tubules, glomeruli and arteries with a cool snapcf camera and autoleveled with Photoshop. Morphometric analysis was performed using MetaVue software. In each image (300 × 900 pixels of proximal tubules and 350 × 350 pixels of glomeruli), the hot pink color area and its intensities, which is representative of interstitial, glomerular and periarterial fibrosis, were quantified.
Transmission Electron Microscopy Methods
Transmission electron microscopy (TEM) images were captured as previously described [16,17]. Briefly, immediately after harvest, 1-2 mm of renal cortical tissues were placed in primary TEM fixative (2.5% of glutaraldehyde and 2.5% of paraformaldehyde). Then, the tissues were transferred into secondary fixative on a rocker overnight, embedded in plastic, the plastic was polymerized at 60°C for 24 h. Ultrathin sections (85 nm) were then stained with 5% of uranyl acetate and Sato's Triple lead citrate. A Jem 1400-EX transmission electron microscope (Joel Ltd., Tokyo, Japan) was utilized to capture the images. Three proximal tubule cells (PTCs) per rat were evaluated with five 10-k and 60-k images. To maintain uniformity, we examined S-1 segments of the PTCs with identifiable microvilli that were immediately adjacent to the glomeruli.
Reactive Oxygen Species Formation
Accumulation of reactive oxygen species (ROS) in kidney tissue was measured by a lucigenin-enhanced chemiluminescence assay. Briefly, kidney tissues were homogenized and centrifuged for 1 h at 100,000 g, and supernatants (whole homogenate) were then removed and placed on ice. Whole homogenate (100 μl) was added to 1.4 ml of 50 mM of phosphate (KH2PO4) buffer (150 mM of sucrose, 1 mM of EGTA, 5 μM of lucigenin, and 100 of μM NADPH; pH 7.0) in dark-adapt counting vials. After dark adaptation, samples were counted on a luminometer, and all counts were averaged. Samples were then normalized to total protein in the whole homogenate.
Quantification of 3-Nitrotyrosine
3-Nitrotyrosine (3-NT) was quantified as previously described [16,17]. Briefly, 5 µm of paraffin-embedded kidney sections from different treatments were dewaxed, rehydrated, antigen retrieved and incubated overnight with 1:150 of primary rabbit polyclonal anti-3-NT antibody (Millipore). Sections were then washed and incubated for 30 min with secondary antibodies, biotinylated anti-rabbit, and streptavidin-horseradish peroxidase. After several rinses with distilled water, diaminobenzidine was applied for 7 min, and sections were again rinsed and stained with hematoxylin for 90 s, dehydrated, and mounted with Permount. The slides were viewed under a bright-field (Nikon 50i) microscope (Nikon, Tokyo, Japan) and 40× images were captured with a Cool SNAP cf camera (Roper Scientific, Trenton, N.J., USA). The images were analyzed by MetaVue (Molecular Devices, Sunnyvale, Calif., USA), and the intensity of brown color which is indicative of 3-NT formation was quantified as gray scale intensity.
Immunohistochemistry
Harvested kidney tissue was prepared as previously described [16,17]. Briefly, rehydrated paraffin-embedded sections were blocked in 5% of BSA, 5% of donkey serum and 0.01% of sodium azide in HEPES buffer for 4 h in a humidity chamber. Following a brief rinse, sections were incubated with 1:100 of rabbit anti-TGF-β1 (Abcam), overnight in humidity chambers at room temperature. Then, the sections were thoroughly washed with HEPES wash buffer and incubated with 1:300 of appropriate secondary antibodies, Alexa fluor donkey anti-mouse, rabbit, goat and rat (Invitrogen) for 4 h, according to the host of the primary antibodies. All primary and secondary antibodies were diluted in a 10-fold diluted blocking agent. After washing, the sections were mounted with Mowiol and were examined under a biphoton laser scanning confocal microscope; the images were captured with the LSM imaging system. Signal intensities were analyzed with MetaVue.
Statistical Analysis
Statistical analyses were performed using Sigma Plot 12.0 (Systat Software, San Jose, Calif., USA) software. Two-way ANOVA and Bonferroni's post hoc test were used as appropriate. All values are expressed as means ± standard error (SE).
Results
Experimental Measures in Female Transgenic Ren2 Rats
In our prior report on the same young transgenic female Ren2 rats, 4% of salt induced diastolic dysfunction accompanied by fibrosis and oxidant stress without changes in systolic pressure [16]. As a marker for kidney function, we then determined proteinuria as a marker for kidney function via a protein:creatinine ratio and observed an increase in proteinuria in the Ren2 rats that was augmented in the presence of a 4%-salt diet compared to controls (fig. 1).
Fig. 1.
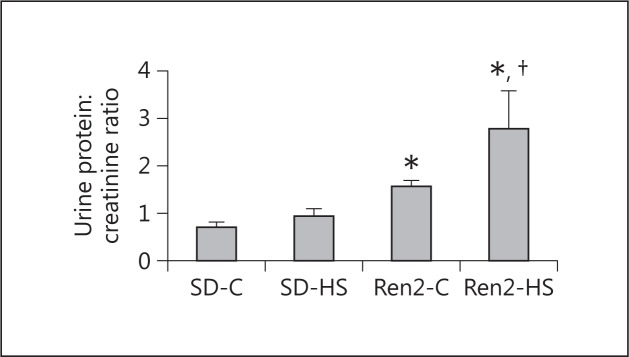
Salt induces protein excretion in female transgenic Ren2 rats. Proteinuria as determined by the urine protein:creatinine ratio is shown. Values are means ± SE. * p < 0.05 when compared to SD controls; † p < 0.05 when compared to Ren2 controls.
Salt-Induced Fibrosis in Female Transgenic Ren2 Rats
The diastolic dysfunction observed in our prior report in response to salt was accompanied by pericapillary fibrosis and alterations in collagen content. Here again similar to the myocardial alterations, 4% of salt induced increases in kidney tissue fibrosis (fig. 2a, b). There were increases in periarterial (fig. 2a) and interstitial fibrosis (fig. 2b) in both SD and Ren2 rats in response to 4% of salt.
Fig. 2.

Salt induces kidney periarterial and interstitial fibrosis in female transgenic Ren2 rats. Representative bright field images of immunostaining with picrosirius red of a periarterial fibrosis and b interstitial fibrosis with corresponding measures of intensity to the right. Values are means ± SE. * p < 0.05 when compared to SD controls; † p < 0.05 when compared to Ren2 controls. Scale bars = 50 µm.
Salt-Induced Ultrastructural Remodeling in Female Transgenic Ren2 Rats
Progressive kidney disease is characterized functionally by increases in proteinuria and morphologically by tubulointerstitial fibrosis. In the earlier stages, proximal tubule promotion of tubulointerstitial fibrosis is characterized by EMT wherein ultrastructural loss of PTC basal polarity and basement membrane thickening occurs. Utilizing TEM, we observed a loss of PTC polarity in female Ren2 rats fed a 4%-salt diet compared to other groups manifested by loss of mitochondria elongation as well as elongated plasma membrane canalicular infoldings that occurred in parallel with basement membrane thickening (fig. 3).
Fig. 3.

Salt induces ultrastructural remodeling of PTCs in female transgenic Ren2 rats. There is a similar morphology of the S1 basilar region of the proximal tubules in control SD rats (SD-C) as well as in Ren2 (Ren2-C) rats with elongation of mitochondria and canalicular infoldings (arrows). Salt had little effect in the SD; however, in the Ren2 (Ren2-HS), we observed increases in basement membrane thickening (BM), loss of mitochondrial elongation, and attenuation of canalicular infoldings compared to the other groups. Scale bars = 1 µM; magnification ×2,000.
Salt Does Not Induce Oxidant Stress but It Induces TGF-β in Female Transgenic Ren2 Rats
Oxidant stress is one mechanism that contributes to kidney tissue remodeling and fibrosis. Both reactive oxygen and nitrogen species contribute to oxidant stress. In this context, there were increases in both ROS generation and 3-NT content, a marker for peroxynitrite formation, in the Ren2 compared to SD controls (fig. 4a, b). In our prior report [16] on salt-fed Ren2 rats, there were further increases in fibrosis that occurred with oxidant stress. However, 4% of salt had no effect on further increases in oxidant markers in the Ren2 kidney yet there was a trend observed in the SD. This led us to explore another critical mechanism of fibrosis in the kidney. TGF-β1 is known to induce Ang II-dependent fibrosis and play a role in EMT [18]. In this regard, similar to our observed increase in periarterial and interstitial fibrosis, there were increases in TGF-β1 in response to 4% of salt in both SD and Ren2 rats (fig. 5).
Fig. 4.

Salt does not induce ROS generation and 3-NT formation in female transgenic Ren2 rats. a ROS generation as determined by chemiluminescence. b Representative sections for 3-NT, a marker of peroxynitrite (ONOO−) formation with corresponding measures of intensity. RLUs = Relative light units. Values are means ± SE. * p < 0.05 when compared to SD controls. Scale bars = 50 µm.
Fig. 5.

Salt induces TGFβ-1 in female transgenic Ren2 rats. Representative images of immunohistochemical analysis with corresponding measures of intensity to the right. Values are means ± SE. * p < 0.05 when compared to SD controls; † p < 0.05 when compared to Ren2 controls. Scale bars = 50 µm.
Discussion
In our current report, we demonstrate that excess salt promotes functional and structural changes in kidney function in young, nonobese transgenic female Ren2 rats that overexpress tissue Ang II and circulating aldosterone. In this regard, 4% of salt over 3 weeks induced increases in proteinuria along with periarterial and interstitial fibrosis and ultrastructural remodeling of the PTC in the Ren2 rats without significant increases in systolic pressure as reported previously in the same animals [16]. Of note, these findings were accompanied by increases in TGF-β1 but not measures of oxidant stress (e.g. ROS formation and 3-NT content). This would be contrary to our observations in the heart [16] wherein 4% of salt induced functional (diastolic dysfunction) and structural (fibrosis) changes that were elicited by increases in oxidant stress. However, our findings support the notion that preconditioning by the RAAS potentiates the actions of salt on tissue fibrosis that have been observed by our group and others [8,13,16]. Our data further suggest that female Ren2 rats fed a 4%-salt diet preconditioned by overactivation of the RAAS overcome the protective impact of gender on kidney injury and kidney disease progression.
Human cohort studies utilizing measures such as estimated glomerular filtration rate and proteinuria support the concept that the protective effect of gender on kidney disease progression is lost in the presence of diabetes. In recent years, there has been an increased interest in exploring this discrepant effect of gender on kidney function and proteinuria under conditions that recreate the diabetic state. In this context, data would indicate that ovariectomy in rodents removes the protective effect of gender on proteinuria suggesting a clear role for sex hormones in kidney function [19]. Our observation that in this female transgenic model increases in proteinuria occurred following a diet high in salt, such as in our Western diet, overcomes this protective effect of gender is novel. These findings further support that dietary factors may recapitulate the loss of gender effect in the presence of diabetes. Our use of the transgenic female Ren2 rat suggests that salt requires excess Ang II and/or aldosterone to precipitate impaired handling of protein. Previous work in this area suggests that increases in proteinuria under conditions of Ang II infusion require 8% of salt intake over 2 weeks [20], while in congenic Ren2: Lewis rats treatment with an agonist of the G protein-coupled estrogen receptor 30 improved proteinuria under a 4%-salt diet [21]. Data from this same study also suggests a higher level of immunostaining of G protein-coupled estrogen receptor 30 in the proximal tubule in this model of excess tissue Ang II, which would support a role for estrogen in Ang II-dependent proximal tubule handling of protein.
In addition to the Ang II-dependent effects on proximal tubule handling of protein, there has been avid interest on the impact of excess aldosterone on proximal tubule function. There is sufficient data to support that aldosterone has a proinflammatory, pro-oxidant effect on interstitial fibrosis in the heart and kidney [22,23]. It is clear that, based on a number of studies, excess salt is obligatory in aldosterone-dependent fibrosis [24]. Yet, there is little understanding of the role that gender may have on aldosterone-/salt-dependent effects. Our novel data suggest that in this female transgenic model of excess circulating aldosterone, excess salt induces kidney periarterial and interstitial fibrosis. There is limited data in this area; however, one study would suggest that drospirenone, a progestin with known mineralocorticoid antagonist function, prevented kidney hypertrophy, increased blood pressure and sodium retention [25]. In this same study, drospirenone along with spironolactone also increased kidney tissue Ang II type 2 receptor expression and improved aldosterone-induced suppression of serum Ang II levels. Considering our data, these findings suggest not only a role for a gender influence on aldosterone-/salt-dependent kidney injury but also that Ang II actions on kidney function require the actions of the mineralocorticoid receptor. In this context, data from one group suggests that despite aldosterone depletion antagonism of the mineralocorticoid receptor improved fibrotic markers in the kidney [26]. These data, in concert with our current observation, suggest that Ang II/salt dependence may also require aldosterone to elicit fibrosis.
Tubulointerstitial fibrosis is the final convergent mechanism that leads to progressive kidney disease and ultimately end-stage renal disease. The mechanisms by which this occurs have been an area of active interest for decades. Tubulointerstitial fibrosis is characterized by accumulation of extracellular matrix in the renal interstitium along with ultrastructural remodeling and loss of cellular architecture [27,28]. Recent evidence would suggest that this remodeling occurs through a process called EMT [18,29]. Unique to our report is that the evidence of ultrastructural remodeling induced by salt is consistent with EMT and consequent fibrosis. EMT is characterized by loss of epithelial cell polarity, loss of cell-cell adhesion and architecture and migration to a mesenchymal phenotype. Our observation that salt-induced ultrastructural remodeling changes consistent with loss of proximal tubule basal polarity, canalicular infoldings, and mitochondrial elongation (mitochondrial fragmentation) along with basement membrane thickness in female Ren2 rats would be a novel finding indicating a potential role for gender in EMT and tubulointerstitial fibrosis. In conjunction with EMT, there has been recent interest in endothelial-mesenchymal transition (EndMT) as either an initiator of EMT or a potential collaborator with EMT in interstitial fibrosis [30]. EndMT is a process wherein endothelial cells contribute to fibroblast migration and fibrosis. Our findings of periarterial fibrosis together with our ultrastructural observations suggest that EndMT and EMT may occur in parallel. The fact that we observed this process in female Ren2 rats fed a salt diet is novel and deserves future investigation. Although our findings are structural in nature and provide little evidence towards changes in the transcriptome, they are particularly noteworthy.
It has been previously documented that Ang II-dependent signaling through the Ang II type 1 receptor or aldosterone activation of the mineralocorticoid receptor combined with salt contributes to inflammation, fibrocyte activation, collagen and extracellular protein synthesis and fibrosis through generation of ROS [8,13,23,26,31]. Our findings that indices of oxidant stress, such as ROS formation and 3-NT content, were increased in the Ren2 rat are consistent with this notion, and our observation that this occurred in females extends work in this area described by the groups mentioned above. However, the lack of a salt-induced effect on oxidant markers in the female Ren2 is of significant interest. In this context, signaling through TGF-β1 has been shown to induce tubulointerstitial fibrosis [6,7]. Most of the evidence support that TGF-β1 synthesis of extracellular matrix, collagen and fibroblast formation occur through direct actions of Ang II on the Ang II type 1 receptor [32] or aldosterone action on the mineralocorticoid receptor [33]. The collective actions of Ang II and aldosterone appear to require salt [33,34,35], and there is work to suggest that endothelial production of TGF-β1 stimulates EMT [36,37]. Thereby, our finding that 4% of salt increased TGF-β1 production along with associated periarterial and interstitial fibrosis in female SD and Ren2 rats complements prior work in this area and extends this work to suggest that there may be a role for gender.
In summary, data from our current investigation denote that salt induces proteinuria and fibrosis without changes in systolic pressure in young female nonobese insulin-resistant Ren2 rats [12,16]. Our data suggest for the first time an important role for gender in processes such as EMT or even EndMT. The observation that these findings occurred in a model of RAAS overactivation support a dual role for both Ang II and aldosterone in salt-induced fibrosis. Our data further suggest that females fed a diet high in salt preconditioned by overactivation of the RAAS overcome the protective impact of gender on kidney injury and kidney disease progression.
Disclosure Statement
The authors have no conflicts of interest to disclose.
Acknowledgements
Female transgenic Ren2 rats and Sprague-Dawley controls were kindly provided by the Transgenic Core Facility at the Wake Forest University School of Medicine, Winston-Salem, N.C., USA (supported in part by NIH grant HL-51952 to C.M.F.). The authors wish to acknowledge the Molecular Cytology core facility and the Electron Microscope core facility for their help and preparation of transmission electron micrographs. Special thanks go to Nathan Rehmer for administrative and technical support.
This research was supported by NIH grants HL73101 and HL1079100 (J.R.S.) and AG040638 (A.T.W.-C.), the Veterans Affairs Merit System (0018) (J.R.S.), CDA-2 (A.T.W.-C.), and the American Society of Nephrology – Association of Specialty Professors (ASN-ASP) Development Grant in Geriatric Nephrology (A.T.W.-C.) are supported by a T. Franklin Williams Scholarship Award, funding provided by Atlantic Philanthropies Inc., the John A. Hartford Foundation.
References
- 1.Iseki K. Gender differences in chronic kidney disease. Kidney Int. 2008;74:415–417. doi: 10.1038/ki.2008.261. [DOI] [PubMed] [Google Scholar]
- 2.Neugarten J, Acharya A, Silbiger SR. Effect of gender on the progression of non-diabetic renal disease: a meta-analysis. Am Soc Nephrol. 2000;11:319–329. doi: 10.1681/ASN.V112319. [DOI] [PubMed] [Google Scholar]
- 3.Stanton RC, King GL. A complex interplay of factors causes diabetic nephropathy. Metabolism. 2011;60:591–593. doi: 10.1016/j.metabol.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Susic D, Frohlich ED. Hypertensive cardiovascular and renal disease and target organ damage: lessons from animal models. Cardiorenal Med. 2011;1:139–146. doi: 10.1159/000329334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.KDOQI Clinical Practice Guideline for Diabetes and CKD 2012 Update. Am J Kidney Dis. 2012;60:850–886. doi: 10.1053/j.ajkd.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int. 2006;70:1914–1919. doi: 10.1038/sj.ki.5001846. [DOI] [PubMed] [Google Scholar]
- 7.Hellmich B, Schellner M, Schatz H, Pfeiffer A. Activation of transforming growth factor-beta1 in diabetic kidney disease. Metabolism. 2000;49:353–359. doi: 10.1016/s0026-0495(00)90264-6. [DOI] [PubMed] [Google Scholar]
- 8.Sun GP, Kohno M, Guo P, et al. Involvements of Rho-kinase and TGF-beta pathways in aldosterone-induced renal injury. J Am Soc Nephrol. 2006;17:2193–2201. doi: 10.1681/ASN.2005121375. [DOI] [PubMed] [Google Scholar]
- 9.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 10.Bhaskaran M, Reddy K, Radhakrishanan N, Franki N, Ding G, Singhal PC. Angiotensin II induces apoptosis in renal proximal tubular cells. Am J Physiol Renal Physiol. 2003;284:F955–F965. doi: 10.1152/ajprenal.00246.2002. [DOI] [PubMed] [Google Scholar]
- 11.Komukai K, Mochizuki S, Yoshimura M. Gender and the renin-angiotensin-aldosterone system. Fundam Clin Pharmacol. 2010;24:687–698. doi: 10.1111/j.1472-8206.2010.00854.x. [DOI] [PubMed] [Google Scholar]
- 12.Johnson MS, DeMarco VG, Heesch CM, et al. Sex differences in baroreflex sensitivity, heart rate variability, and end organ damage in the TGR(mRen2)27 rat. Am J Physiol Heart Circ Physiol. 2011;301:H1540–H1550. doi: 10.1152/ajpheart.00593.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen JA, Lindsey SH, Pirro NT, et al. Influence of estrogen depletion and salt loading on renal angiotensinogen expression in the mRen(2). Lewis strain. Am J Physiol Renal Physiol. 2010;299:F35–F42. doi: 10.1152/ajprenal.00138.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones-Burton C, Mishra SI, et al. An in-depth review of the evidence linking dietary salt intake and progression of chronic kidney disease. Am J Nephrol. 2006;26:268–275. doi: 10.1159/000093833. [DOI] [PubMed] [Google Scholar]
- 15.Blendea MC, Jacobs D, Stump CS, et al. Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression. Am J Physiol Endocrinol Metab. 2005;288:E353–E359. doi: 10.1152/ajpendo.00402.2004. [DOI] [PubMed] [Google Scholar]
- 16.Whaley-Connell AT, Habibi J, Aroor A, et al. Salt loading exacerbates diastolic dysfunction and cardiac remodeling in young female Ren2 rats. Metabolism. 2013;62:1761–1771. doi: 10.1016/j.metabol.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Habibi J, Hayden MR, Sowers JR, et al. Nebivolol attenuates redox-sensitive glomerular and tubular mediated proteinuria in obese rats. Endocrinology. 2011;152:659–668. doi: 10.1210/en.2010-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121:468–474. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaleyeva LM, Pendergrass KD, Pirro NT, et al. Ovariectomy is protective against renal injury in the high-salt-fed older mRen2. Lewis rat. Am J Physiol Heart Circ Physiol. 2007;293:H2064–H2071. doi: 10.1152/ajpheart.00427.2007. [DOI] [PubMed] [Google Scholar]
- 20.Rands VF, Seth DM, Kobori H, Prieto MC. Sexual dimorphism in urinary angiotensinogen excretion during chronic angiotensin II-salt hypertension. Gend Med. 2012;9:207–218. doi: 10.1016/j.genm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindsey SH, Yamaleyeva LM, Brosnihan KB, et al. Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt-sensitive female mRen2. Lewis rat. Hypertension. 2011;58:665–671. doi: 10.1161/HYPERTENSIONAHA.111.175174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aroor AR, McKarns S, Demarco VG, et al. Maladaptive immune and inflammatory pathways lead to cardiovascular insulin resistance. Metabolism. 2013;62:1543–1552. doi: 10.1016/j.metabol.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pimenta E, Calhoun DA. Aldosterone, dietary salt, and renal disease. Hypertension. 2006;48:209–210. doi: 10.1161/01.HYP.0000230482.82239.a0. [DOI] [PubMed] [Google Scholar]
- 24.Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol. 2013;9:459–469. doi: 10.1038/nrneph.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arias-Loza PA, Muehlfelder M, Elmore SA, et al. Differential effects of 17beta-estradiol and of synthetic progestins on aldosterone-salt-induced kidney disease. Toxicol Pathol. 2009;37:969–982. doi: 10.1177/0192623309350475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luther JM, Luo P, Wang Z, et al. Aldosterone deficiency and mineralocorticoid receptor antagonism prevent angiotensin II-induced cardiac, renal, and vascular injury. Kidney Int. 2012;82:643–651. doi: 10.1038/ki.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwano M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. Curr Opin Nephrol Hypertens. 2004;13:279–284. doi: 10.1097/00041552-200405000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Rodríguez-Iturbe B, García García G. The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron Clin Pract. 2010;116:c81–c88. doi: 10.1159/000314656. [DOI] [PubMed] [Google Scholar]
- 29.Hewitson TD. Renal tubulointerstitial fibrosis: common but never simple. Am J Physiol Renal Physiol. 2009;296:F1239–F1244. doi: 10.1152/ajprenal.90521.2008. [DOI] [PubMed] [Google Scholar]
- 30.Zeisberg EM, Potenta SE, Sugimoto H, et al. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lastra G, Santos FR, Hooshmand P, Hooshmand P, Mugerfeld I, Aroor AR, Demarco VG, Sowers JR, Henriksen EJ. The novel angiotensin II receptor blocker azilsartan medoxomil ameliorates insulin resistance induced by chronic angiotensin II treatment in tat skeletal muscle. Cardiorenal Med. 2013;3:154–164. doi: 10.1159/000353155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolf G, Mueller E, Stahl RAK, Ziyadeh FN. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor. J Clin Invest. 1993;92:1366–1372. doi: 10.1172/JCI116710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hovater MB, Sanders PW. Effect of dietary salt on regulation of TGF-β in the kidney. Semin Nephrol. 2012;32:269–276. doi: 10.1016/j.semnephrol.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juknevicius I, Segal Y, Kren S, et al. Effect of aldosterone on renal transforming growth factor-beta. Am J Physiol Renal Physiol. 2004;286:F1059–F1062. doi: 10.1152/ajprenal.00202.2003. [DOI] [PubMed] [Google Scholar]
- 35.Kim S, Ohta K, Hamaguchi A, et al. Role of angiotensin II in renal injury of deoxycorticosterone acetate-salt hypertensive rats. Hypertension. 1994;24:195–204. doi: 10.1161/01.hyp.24.2.195. [DOI] [PubMed] [Google Scholar]
- 36.Hills CE, Squires PE. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am J Nephrol. 2010;31:68–74. doi: 10.1159/000256659. [DOI] [PubMed] [Google Scholar]
- 37.Hayden MR, Sowers KM, Pulakat L, Joginpally T, Krueger B, Whaley-Connell A, Sowers JR. Possible mechanisms of local tissue renin-sngiotensin system activation in the cardiorenal metabolic syndrome and type 2 diabetes mellitus. Cardiorenal Med. 2011;1:193–210. doi: 10.1159/000329926. [DOI] [PMC free article] [PubMed] [Google Scholar]