

Abstract
Alcohol is a major cause of chronic pancreatitis. About 5% of alcoholics will ever suffer from pancreatitis, suggesting that additional co-factors are required to trigger an overt disease. Experimental work has implicated lipopolysaccharide, from gut-derived bacteria, as a potential co-factor of alcoholic pancreatitis. This review discusses the effects of alcohol on the gut flora, the gut barrier, the liver-and the pancreas and proposes potential interventional strategies. A better understanding of the interaction between the gut, the liver and the pancreas may provide valuable insight into the pathophysiology of alcoholic pancreatitis.
Keywords: Alcohol, Pancreatitis, Fibrosis, Bacteria, Endotoxin, Lipopolysaccharide
Core tip: There is now clear clinical and experimental evidence that bacteria and bacterial products (such as endotoxin) are associated with complications of pancreatitis. Furthermore, results of animal studies support the concept that bacterial endotoxin is an important factor in the initiation and progression of alcoholic pancreatitis.
INTRODUCTION
Chronic alcohol consumption is a known cause of injury to several organs, most commonly the liver and the pancreas, but also to the heart, lungs and brain. However, it is well understood that only a minority of alcoholics will ever develop clinically overt pancreatic or liver damage and even fewer numbers will develop clinically overt disease in both organs simultaneously although subclinical damage to both organs has been reported to coexist[1]. The fact that only some alcoholics appear to be susceptible to clinical pancreatitis or hepatitis has led to a concerted search for additional trigger/initiating factors for alcohol-induced organ damage.
Over the past two decades clinical and experimental studies have demonstrated that endotoxin lipopolysaccharide (LPS), from the bacterial wall of gram negative bacteria of the human gut, plays a central role in the initiation and progression of alcoholic liver disease[2]. This was initially based on clinical observations of elevated plasma endotoxin concentrations in alcoholics with and without liver disease[3,4]. Experimental evidence in support of the association of endotoxin and liver disease in humans was subsequently provided by animal studies demonstrating that alcohol-fed rats challenged with LPS developed hepatic lesions resembling alcoholic hepatitis in humans[5,6]. Conversely, targeted disruption of the LPS receptor toll like receptor 4 (TLR4) in alcohol-fed animals protected against liver injury[7].
Reports of increased endotoxinemia in pancreatitis emerged a decade later. Several studies have linked the degree of endotoxinemia to the severity and prognosis of acute pancreatitis, regardless of its aetiology[8,9] and the impact of endotoxinemia on multiple organ system failure, in particular pancreatitis-associated lung disease has been corroborated by animal studies[10]. However, it remained elusive whether endotoxinemia was a cause or a consequence of pancreatitis, or both. It has only recently been shown that endotoxin initiates pancreatic necro-inflammation in alcohol-fed rodents[11,12] and promotes pancreatic fibrosis[12].
In healthy subjects, small amounts of endotoxin translocate from the gut lumen to the bloodstream and are naturally cleared by the reticulo-endothelial system. Under the influence of alcohol, bacteria proliferate in the small intestine[13,14], intestinal permeability is increased[15,16], while endotoxin clearance by the reticulo-endothelial system-in particular Kupffer cells in the liver - is diminished[17]. As a result, excess endotoxin is available in the blood stream and exerts its harmful effects on various organs.
This review aims to summarise the mechanisms underlying increased endotoxinemia in alcoholics, describes the role of endotoxin both as an initiating and aggravating factor of pancreatitis and attempts to define a role for the liver as a mediator in pancreatic end-organ damage.
ALCOHOL AND THE GUT FLORA
A human being harbours up to 500 different bacterial species[18], the overall bacterial cell count being 10 times more abundant than the number of eukaryotic cells in the body[19]. The combination of species-which is established during the first year of life and shaped by host genotype[20] as well as dietary factors-varies from individual to individual[21]. Moreover, there is evidence indicating that certain strains of bacteria may be unique to their host[22]. Bacterial concentrations are lowest in the upper gastrointestinal tract due to gastric acid, biliary and pancreatic secretion while the highest density of bacteria is found in the colon. In healthy humans, the gut flora prevents the growth of potential injurious bacteria[18,23], exerts metabolic activities such as the fermentation of non-digestible carbohydrates[24] or vitamin synthesis[25] and plays a role in intestinal cell growth and differentiation[26]. Several factors may influence bacterial luminal content. These include altered gut motility[27], drugs, in particular antibiotics[28] and dietary factors such as alcohol.
Alcohol has been shown to alter the jejunal microflora, since almost 50% of alcoholics with documented recent ethanol abuse displayed an increase in total number of bacteria most of which originated from the faecal flora[13]. These data were confirmed in duodenal juice samples obtained by oesogastroduodenoscopy[14] as well as H2-breath tests, as a surrogate marker of bacterial proliferation in the proximal gut, in alcoholic subjects[29]. The mechanisms underlying bacterial overgrowth in alcoholism are unknown, but reduction of orocaecal transit time observed in chronic alcoholics[30,31] may offer a partial explanation. It is noteworthy, that alcohol gavage in rodents for 10 wk has the capacity to alter the composition of colonic bacteria[32].
Interestingly, certain bacteria of the gut flora have the capacity to metabolise alcohol to acetaldehyde[33,34]. In alcohol-fed rats, ethanol metabolism by colonic bacteria could be suppressed by ciprofloxacin[35] or a combination of ampicillin and neomycin[36]. In a similar animal model, administration of metronidazole increased alcohol dehydrogenase-containing bacteria and hence colonic acetaldehyde content[37]. While acetaldehyde has been measured in the rodent colon[36] and human gut bacteria have the capacity to metabolise ethanol, there is, to date, no report on acetaldehyde content of the human colon in alcoholics. Nonetheless, the above studies suggest that it would not be unreasonable to implicate acetaldehyde, as the compound that mediates most of the toxic effects of ethanol.
ALCOHOL AND GUT PERMEABILITY
In order for bacteria or bacterial products such as endotoxin to pass into the bloodstream and exert their systemic effects, they are required to cross the gut barrier. In its physiological state, the gut represents an effective barrier, made of a single continuous cell layer from the stomach to the rectum. The cells are sealed together by two sets of highly complex junctions, the more apical tight junction and the adherens junction. Physiologically, tight junctions may allow the passage of small molecules up to a molecular weight of 2000 Da but prevents the translocation of larger molecules, in particular bacterial products or bacteria[38]. In addition to this mechanical barrier, passage of bacteria or bacterial products is prevented by mucus, immunoglobulins, defensins and other antimicrobial products produced by the gut.
Intestinal permeability can be measured non-invasively using oral probes such as ethylene glycol polymers of varying molecular sizes, oligosaccharides (e.g., lactulose), monosaccharides (mannitol) and radiolabeled chelates such as chromium-ethylenediaminetetraacetic acid (Cr-EDTA). All these compounds are poorly absorbed by the normal bowel mucosa and display absent or negligible metabolism. Hence, increased urinary excretion correlates with increased intestinal permeability. It is now acknowledged that the probes are absorbed via the paracellular route, implying that competence of the gut barrier depends on the integrity of intercellular junctions[39,40].
Several studies have addressed the question whether alcohol increases gut permeability. Early studies with rats chronically administered alcohol revealed increased permeability to macromolecules such as hemoglobin with a known molecular weight of 17 kDa[41] and horseradish peroxidase with a molecular weight of 44 kDa[42]. Permeability to smaller molecules also appears to be increased in rodents upon ethanol administration as exemplified by increased lactulose/mannitol ratio. Increased absorption of 51Cr-EDTA, a small molecule of 340 Da, was also observed in chronic alcoholics[42]. An increase in absorption of a molecule of similar size (PEG 400) was reported when alcohol was administered to volunteers with no history of chronic ethanol abuse[16]. The latter data failed to be confirmed by Parlesak et al[43] who did not observe a difference in the absorption of polyethylen glycol (PEG) 400 when chronic alcoholics were compared to healthy subjects. In the same study, however, permeability to larger molecules of polyethylene glycol (PEG 1500, 4000 and 10000) was significantly enhanced and the permeability to PEG 10000 in particular was 10-fold higher in alcoholics. Taken together there is experimental and clinical evidence that gut permeability is enhanced by acute and chronic ethanol administration. Permeability seems to be increased for molecules of higher molecular weight (from 1000 Da to at least 44 kDa), which is of particular relevance to the translocation of gut derived bacterial endotoxin, a large compound with a known molecular weight of 40 kDa, as a putative initiating and aggravating factor of alcohol-induced organ damage.
In order to explain increased gut permeability by alcohol, various morphological and molecular studies have been undertaken. There is evidence that alcohol exerts direct toxic effects on the gut mucosa. In an observational study by Gottfried et al[44], seven alcoholic subjects with a previously unremarkable oesogastroduodenoscopy were administered 1 g/kg body weight alcohol (35% w/v). Biopsy specimens taken during oesogastroduodenoscopy performed 3 h after alcohol exposure demonstrated transient focal subepithelial hemorrhage which disappeared within 3 d. These observations were corroborated by experimental data in rodents and dogs[45,46]. Studies of histological alterations in patients chronically abusing alcohol have yielded conflicting results since both histological alterations and normal mucosal structure have been described[47]. This may be related to the fact that alcohol-induced mucosal lesions are short-lived due to rapid regeneration of epithelial cells (in the study reporting normal mucosal structure, endoscopies were performed 3-14 d after alcohol withdrawal). At the molecular level, different effects of ethanol on interepithelial junctions in the gut have been described.
Ethanol at high doses has been reported to lead to increased gut permeability via direct action on tight junctions. Ma et al[48] measured epithelial resistance and paracellular permeability of the human adenocarcinoma cell line Caco-2 exposed to ethanol. At ethanol concentration ranging from 1% to 10% a dose-dependent drop in electrical resistance paralleled by an increase in permeability was observed. Ethanol produced a disruption of the tight junction protein ZO-1 as well as disassembly of cytoskeletal proteins such as actin and myosin. These changes proved reversible upon ethanol withdrawal. However, ethanol concentrations of 1% or above are only encountered in the duodenum/jejunum where concentrations of up to 5% have been reported[49], while ethanol concentrations in the ileum and colon tend to be much lower (0.2%-0.25%). This would entail that most of translocation of bacteria or bacterial products occurs in the upper gastrointestinal tract.
As mentioned above, human colonic bacteria have the capacity to metabolise alcohol to acetaldehyde[33,50] via bacterial alcohol dehydrogenase. Accordingly, colonic acetaldehyde concentrations in the millimolar range have been observed in rats[51] and piglets[52]. Acetaldehyde concentrations of 0.1-0.6 mmol/L led to a disruption of tight junctions and adherens junction via tyrosine phosphorylation of their main components[53].
In summary, there is substantial evidence that alcohol increases gut permeability to large molecules of the size of endotoxin and these effects may be due to a direct toxic effect on the mucosa of the proximal gut as well as molecular modifications at the level of interendothelial junctions. Likewise, acetaldehyde, as a result of alcohol metabolism by colonic bacteria, has the capacity to disrupt epithelial junctions, suggesting that the increased serum endotoxin concentrations observed in alcoholics may also be of colonic origin.
BACTERIA AND LPS IN PANCREATITIS
In the Western society, alcohol represents 70%-80% of cases of chronic pancreatitis. As stated earlier, experimental evidence suggests that bacterial endotoxin is an initiating factor for alcoholic pancreatitis[11,12]. In addition, bacterial translocation or the passage of bacterial products such as endotoxin into the systemic circulation appears to play a primary role in systemic spread, including multiple organ system failure and prognosis of the disease[54]. While endotoxin may be a key player at both ends of the disease spectrum, i.e., as an initiating and aggravating factor of pancreatitis, the mechanisms leading to its increased presence in the blood may not be the same. In this chapter, both situations will be considered separately. The question as to whether bacteria or bacterial products (LPS) translocate will be addressed first.
Sepsis, a consequence of infected pancreatic necrosis, accounts for up to 80% of deaths in severe acute pancreatitis[55]. The germs most commonly cultured from infected pancreatic necrosis are gram negative bacilli presumably as a result of increased gut permeability[55,56]. Infection of pancreatic necrosis appears to be an early event occurring within a week after initiation of the disease in more than a quarter of patients undergoing necrosectomy[55,57]. However, the translocation of entire bacteria from the gut to the systemic circulation has not been proven so far in a setting of human acute pancreatitis. Indeed, blood cultures from patients with severe acute pancreatitis are often sterile even with established infected pancreatic necrosis[58]. Ammori et al[54] investigated the presence of bacterial DNA in the systemic circulation of 26 patients with acute pancreatitis. No bacterial DNA was detected in any of the samples. In one patient blood cultures subsequently turned out to be positive for E. Coli. This study suggests that translocation of entire bacteria, as opposed to bacterial products, rarely occurs in acute pancreatitis. However, it has to be noted that the administration of prophylactic antibiotics to 9 of 19 patients with mild attacks and all 7 patients with severe attacks of pancreatitis may have prevented significant bacterial translocation.
Endotoxin is detectable in the majority of patients with established severe acute pancreatitis, in particular in more than 90% of patients dying of the disease[59,60]. Measuring circulating anti-endotoxin antibodies Barclay et al[61] have observed a significant decrease in antibody titres in patients with severe acute pancreatitis compared to patients with mild disease, suggesting higher endotoxin exposure in the former. In a comprehensive study, Ammori et al[8] undertook to measure intestinal barrier function (by measuring intestinal permeability using a PEG probe of 3350 Da) early in the course of acute pancreatitis and to examine the correlation between intestinal permeability, endotoxinaemia and disease severity. Intestinal permeability was significantly increased in patients with severe acute pancreatitis in comparison to mild disease and disease-free controls. Changes in permeability occured early in the course of the disease, before the development of multiple organ system failure. Endotoxinaemia correlated with intestinal permeability and was present more frequently and at higher concentrations in patients with severe disease. Similar observations were made by Windsor et al[9] demonstrating that a significant fall in serum concentrations of immunoglubulin G antiendotoxin core antibodies as a surrogate marker for endotoxemia in patients with acute pancreatitis was predictive of pancreatitis severity and multiple organ system failure.
LPS has also been reported to be a disease modifier in experimental non-alcoholic pancreatitis induced by various treatments. In a rat model of acute pancreatitis induced by the closed duodenal loop procedure[62] disease severity was significantly worsened by endotoxin administration[62]. Pastor et al[63] studied the direct effect of bacterial endotoxin on the course of caerulein-induced acute pancreatitis and pancreatitis-associated lung injury in TLR4 knockout mice and TLR4 sufficient controls. Administration of LPS alone did not induce pancreatitis per se nor did it potentiate the effects of cerulein on the pancreas in either mouse strain. However, there was a significant deterioration of pancreatitis-associated lung injury when LPS was combined with cerulein in wild type mice; lung injury was significantly reduced in TLR4 knockout mice implying that the effect of LPS was mediated via the TLR4 pathway[63]. Surprisingly, targeted deletion of TLR4 and CD14 in mouse models of cerulein- and Arginine-induced pancreatitis without LPS administration, resulted in attenuated pancreatitis and pancreatitis-associated lung injury[64]. The latter study suggests that “endogenous” endotoxin might play a role in the pathophysiology of these models or that LPS receptors play additional roles other than LPS signal transduction in pancreatitis.
The question whether endotoxinemia is an initiating event of alcoholic pancreatitis, similar to alcoholic liver disease has been approached in animal models. As noted earlier, it is well known that only a minority of alcoholics will ever develop acute pancreatitis suggesting that additional factors are required to elicit overt disease. This is evidenced by experimental work in rodents where long-term administration of ethanol did not lead to pancreatitis[65]. Fortunato et al[11] studied the effect of intravenous LPS administration on rats fed a Lieber-de Carli liquid diet with or without alcohol. Using single LPS doses of up to 3 mg/kg body weight, the authors showed a dose-dependent increase in pancreatic lesions, while rats fed alcohol alone did not display significant pancreatic damage. In accordance with the hypothesis whereby repeated attacks of acute pancreatitis lead to chronic disease (necrosis-fibrosis sequence proposed by Ammann et al[66]), Vonlaufen et al[12] showed that repeated weekly injections of endotoxin to alcohol-fed rats led to significant pancreatic fibrosis via a TLR4 mediated effect on pancreatic stellate cells (PSCs), the main effectors of pancreatic fibrosis. Moreover, the presence of TLR4 and its co-receptor CD14 was detected on disease-associated and normal human pancreatic stellate cells[12,67], suggesting that PSCs are a relevant target for endotoxin in human alcoholic pancreatitis.
Taken together, endotoxin (from gut derived bacteria) appears to be an aggravating factor of pancreatitis and associated extra-pancreatic organ damage regardless of aetiology. Furthermore, there is increasing (experimental) evidence that it may play a specific role in the initiation and progression of alcoholic pancreatitis.
THE GUT-LIVER-PANCREAS AXIS
In healthy humans, trace amounts of endotoxin may transiently enter the portal circulation and are cleared by Kupffer cells in the liver. When alcohol is consumed, the detoxifying capacity of the liver seems overwhelmed, since endotoxin is detected in the systemic circulation. In 1987, Bode et al[3] showed for the first time that gut-derived endotoxin is increased in the systemic circulation after acute alcohol consumption by subjects with or without liver damage. The authors evaluated peripheral venous blood endotoxin concentrations in patients with alcoholic and non-alcoholic cirrhosis and in a group of alcoholics with no evidence of chronic liver disease. Increased endotoxin concentrations were found in a significantly larger proportion of patients with alcoholic liver disease (67.3%) than patients with liver disease of non-alcoholic aetiology (45.5%, P < 0.025). Moreover, almost half of all subjects without preexisting liver disease, presenting after a single alcoholic binge, were found to have endotoxin in the blood; importantly, in this group endotoxinemia appeared to be a transient phenomenon with no endotoxin detected after 5-8 d. Further work by the same group confirmed elevated blood endotoxin levels in a significantly higher proportion of patients with alcoholic cirrhosis compared to patients with cirrhosis of a different cause. It is noteworthy, that mean blood endotoxin concentrations were significantly higher in cirrhotics of alcoholic aetiology (19 ± 2.3 vs 12 ± 3.1 pg/mL, P < 0.025)[4].
Early work in patients with cirrhosis has reported toxic effects of alcohol on the reticulo-endothelial system, notably reduced phagocytic and metabolic activity of macrophages[68]. Experimentally, Kupffer cells from alcohol-fed rodents treated in vitro with ethanol at concentrations ranging from 10 to 100 mmol/L (corresponding to alcohol concentrations found in moderate drinkers and severe alcoholics respectively) displayed reduced endotoxin uptake and decreased production of the proinflammatory cytokine tumor necrosis factor alpha (TNF-α), an effect that was dose-dependent[69]. Endotoxin alone activates Kupffer cells by increasing their phagocytic capacity and inducing the production of proinflammatory cytokines (such as TNF-α and interleukin-6 )[70].
Whether concomitant liver disease is a co-factor for alcoholic pancreatitis remains elusive. It is well known that patients with cirrhosis are predisposed to episodes of bacterial infections, including spontaneous bacterial peritonitis with bacteria of gut origin[71,72]. Liver disease impacts on small bowel motility (and potentially bacterial overgrowth), and this effect worsens with increasing severity of liver disease[73]. Experimentally, CCl4-induced cirrhosis resulted in enterocyte oxidative stress, altered enterocyte mitochondrial function, increased lipid peroxidation and altered intestinal transport[74]. Part of the oxidative stress occurring in the enterocyte appears to be related to increased xanthine oxidase activity and increased intestinal permeability, a mechanism that can be blocked experimentally by the administration of xanthine oxidase inhibitors[75]. Accordingly, administration of allopurinol to patients with established cirrhosis efficiently reduced (systemic) oxidant stress, but did not have a significant effect on intestinal permeability[76].
Do alcoholic liver and pancreas disease occur together? A recent study by Yang et al[77] reviewing the epidemiology of alcohol-related pancreatic and liver disease in the United States, has reported that the prevalence of patients discharged with a diagnosis of both acute alcoholic pancreatitis and acute alcoholic hepatitis or both chronic alcoholic pancreatitis and chronic alcoholic liver disease was significantly lower than the prevalence of either disease alone. This is in conflict with necropsy data suggesting that subclinical damage to both organs often coexists[1].
PROPHYLAXIS AND SUPPORTIVE TREATMENT
Alcohol abstinence is the most obvious prophylaxis for alcoholic pancreatitis. Studies suggest that it reduces the incidence of acute attacks and retards clinical progression of the disease[78]. However, this goal is seldom reached and recurrence is common[79] (Figure 1).
Figure 1.
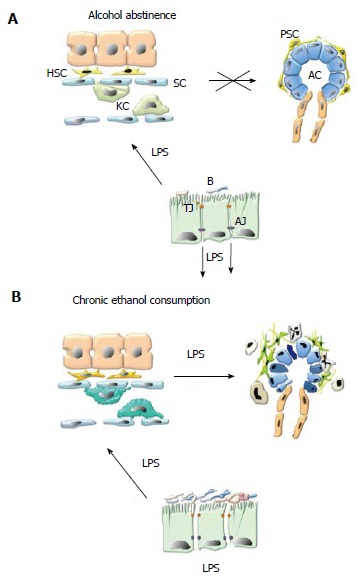
Alcohol and lipopolysaccharide promote pancreatic necroinflammation and fibrosis via pancreatic stellate cell activation. A: Alcohol abstinence. In healthy, non-alcoholic subjects small amounts of lipopolysaccharide (LPS) derived from the membrane of commensal gram negative bacteria (B) cross the gut epithelial barrier at the level of interendothelial junctions. LPS reaches the liver via the portal circulation where it is entirely cleared by Kupffer cells (KC) in the liver sinusoids (S), preventing it from entering the systemic circulation and reaching systemic organs such as the pancreas; B: Chronic ethanol consumption. Chronic alcohol consumption promotes bacterial proliferation in the proximal small bowel, dissociation of interendothelial junctions (by direct toxicity of alcohol and its metabolites) and leads to increased translocation of LPS into the portal circulation. In the liver, alcohol decreases the phagocytic capacity of Kupffer cells. As a result, LPS enters the systemic circulation and exerts its harmful effects on the pancreas. Alcohol and LPS promote pancreatic necroinflammation and fibrosis via PSC activation. TJ: Tight junctions; AJ: Adherens junctions; AC: Acinar cell; PSC: Pancreatic stellate cell.
Since bacteria or bacterial products appear to play a primary role in the initiation, progression and rate of complications of alcoholic pancreatitis, it appears logical to target gut bacteria either within the lumen via bacterial decontamination with nonabsorbable antibiotics or once translocation has occurred, via systemic administration of antibiotics.
Experimental evidence in rodents suggests that selective bacterial decontamination by oral, non absorbable antibiotics significantly reduced the incidence of pancreatic infection[80-82]. However, the application of prophylactic antibiotics in patients with acute pancreatitis has proven ineffective in a large randomized trial comparing the administration of meropenem vs placebo[83]. Another way to influence bacterial luminal content and act on gut barrier integrity may be the application of probiotics (mostly lactobacilli or bifidobacterium strains), that is bacteria which exert protective effects on gut epithelial integrity and prevent colonization by pathogens[84]. However, in a large multicentre randomized controlled trial administration of a cocktail of probiotic bacterial strains (4 lactobacilli and 2 bifidobacteria)[85] within 72 h after onset of symptoms of pancreatitis was of no proven benefit. Moreover, excess mortality in the probiotic group was observed, with one third of deaths related to bowel ischemia. All of these patients presented with early organ failure. In a substudy it became apparent that administration of these particular probiotic bacterial strains in patients with multiple organ failure resulted in increased gut mucosal damage and permeability, as assessed by urinary intestinal fatty acid binding protein IFABP and NOx concentrations, while bacterial translocation was reduced in patients without organ failure[86].
Several animal and human studies have shown that enteral nutrition has a beneficial effect on gut mucosal integrity. In a recent meta-analysis by Petrov et al[87] including 5 randomised controlled trials in patients with severe acute pancreatitis, it was concluded that enteral feeding led to a significant reduction of pancreatic infections, other infectious complications and mortality, but not of organ failure. Another meta-analysis including 8 randomised controlled trials reached similar conclusions but also recorded a significant reduction in organ failure and need for surgical interventions in the total enteral nutrition (TEN) groups as compared to patients receiving total parenteral nutrition[87]. Despite overwhelming evidence in favour of early TEN in a setting of acute pancreatitis, the dogma that the diseased pancreas needs to be “put at rest” still prevails in many centers.
Taken together, early enteral nutrition significantly reduces infectious complications and mortality in patients suffering from acute pancreatitis regardless of aetiology. In contrast, the systematic administration of systemic antibiotics or of probiotics can not be recommended. To date, prophylactic studies aiming at inhibiting gut barrier dysfunction/bacterial translocation in alcoholic subjects are lacking.
CONCLUSION
There is now clear clinical and experimental evidence that bacteria and bacterial products such as endotoxin are associated with complications of pancreatitis. Furthermore, results of animal studies support the concept that bacterial endotoxin is an important factor in the initiation and progression of alcoholic pancreatitis.
Since all alcoholics may be expected to have bacterial translocation, the fact that only a minority develops overt pancreatitis indicates that genetic polymorphism plays a primordial role. Nonetheless, only two candidate genes (carboxylester lipase[88] and chymotrypsin C[89])-explaining a minoritiy of cases of alcoholic pancreatitis-have been identified so far. Additional case-control studies, comparing alcoholics with pancreatitis to alcoholics without pancreatic disease, and targeting genes encoding tight junctional proteins or LPS-receptors are needed to clarify the issue. Moreover, particular attention should be paid to the assessment of the quality of the microbiome in these two populations.
Footnotes
P- Reviewers: Kuan YH, Zhao Y S- Editor: Song XX L- Editor: A E- Editor: Wu HL
References
- 1.Pace A, de Weerth A, Berna M, Hillbricht K, Tsokos M, Bläker M, Pueschel K, Lohse AW. Pancreas and liver injury are associated in individuals with increased alcohol consumption. Clin Gastroenterol Hepatol. 2009;7:1241–1246. doi: 10.1016/j.cgh.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F, Kang YJ, Keshavarzian A, Rao R, Sartor RB, et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol. 2008;42:349–361. doi: 10.1016/j.alcohol.2008.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol. 1987;4:8–14. doi: 10.1016/s0168-8278(87)80003-x. [DOI] [PubMed] [Google Scholar]
- 4.Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol. 1991;12:162–169. doi: 10.1016/0168-8278(91)90933-3. [DOI] [PubMed] [Google Scholar]
- 5.Bhagwandeen BS, Apte M, Manwarring L, Dickeson J. Endotoxin induced hepatic necrosis in rats on an alcohol diet. J Pathol. 1987;152:47–53. doi: 10.1002/path.1711520107. [DOI] [PubMed] [Google Scholar]
- 6.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 7.Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- 8.Ammori BJ, Leeder PC, King RF, Barclay GR, Martin IG, Larvin M, McMahon MJ. Early increase in intestinal permeability in patients with severe acute pancreatitis: correlation with endotoxemia, organ failure, and mortality. J Gastrointest Surg. 1999;3:252–262. doi: 10.1016/s1091-255x(99)80067-5. [DOI] [PubMed] [Google Scholar]
- 9.Windsor JA, Fearon KC, Ross JA, Barclay GR, Smyth E, Poxton I, Garden OJ, Carter DC. Role of serum endotoxin and antiendotoxin core antibody levels in predicting the development of multiple organ failure in acute pancreatitis. Br J Surg. 1993;80:1042–1046. doi: 10.1002/bjs.1800800840. [DOI] [PubMed] [Google Scholar]
- 10.Pastor CM, Rubbia-Brandt L, Hadengue A, Jordan M, Morel P, Frossard JL. Role of macrophage inflammatory peptide-2 in cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. Lab Invest. 2003;83:471–478. doi: 10.1097/01.lab.0000063928.91314.9f. [DOI] [PubMed] [Google Scholar]
- 11.Fortunato F, Deng X, Gates LK, McClain CJ, Bimmler D, Graf R, Whitcomb DC. Pancreatic response to endotoxin after chronic alcohol exposure: switch from apoptosis to necrosis? Am J Physiol Gastrointest Liver Physiol. 2006;290:G232–G241. doi: 10.1152/ajpgi.00040.2005. [DOI] [PubMed] [Google Scholar]
- 12.Vonlaufen A, Xu Z, Daniel B, Kumar RK, Pirola R, Wilson J, Apte MV. Bacterial endotoxin: a trigger factor for alcoholic pancreatitis? Evidence from a novel, physiologically relevant animal model. Gastroenterology. 2007;133:1293–1303. doi: 10.1053/j.gastro.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 13.Bode JC, Bode C, Heidelbach R, Dürr HK, Martini GA. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology. 1984;31:30–34. [PubMed] [Google Scholar]
- 14.Hauge T, Persson J, Danielsson D. Mucosal bacterial growth in the upper gastrointestinal tract in alcoholics (heavy drinkers) Digestion. 1997;58:591–595. doi: 10.1159/000201507. [DOI] [PubMed] [Google Scholar]
- 15.Bjarnason I, Williams P, So A, Zanelli GD, Levi AJ, Gumpel JM, Peters TJ, Ansell B. Intestinal permeability and inflammation in rheumatoid arthritis: effects of non-steroidal anti-inflammatory drugs. Lancet. 1984;2:1171–1174. doi: 10.1016/s0140-6736(84)92739-9. [DOI] [PubMed] [Google Scholar]
- 16.Robinson GM, Orrego H, Israel Y, Devenyi P, Kapur BM. Low-molecular-weight polyethylene glycol as a probe of gastrointestinal permeability after alcohol ingestion. Dig Dis Sci. 1981;26:971–977. doi: 10.1007/BF01314757. [DOI] [PubMed] [Google Scholar]
- 17.Järveläinen HA, Fang C, Ingelman-Sundberg M, Lindros KO. Effect of chronic coadministration of endotoxin and ethanol on rat liver pathology and proinflammatory and anti-inflammatory cytokines. Hepatology. 1999;29:1503–1510. doi: 10.1002/hep.510290508. [DOI] [PubMed] [Google Scholar]
- 18.Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361:512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- 19.Bengmark S. Ecological control of the gastrointestinal tract. The role of probiotic flora. Gut. 1998;42:2–7. doi: 10.1136/gut.42.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7:688–693. doi: 10.1038/sj.embor.7400731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simon GL, Gorbach SL. Intestinal flora in health and disease. Gastroenterology. 1984;86:174–193. [PubMed] [Google Scholar]
- 22.Kimura K, McCartney AL, McConnell MA, Tannock GW. Analysis of fecal populations of bifidobacteria and lactobacilli and investigation of the immunological responses of their human hosts to the predominant strains. Appl Environ Microbiol. 1997;63:3394–3398. doi: 10.1128/aem.63.9.3394-3398.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guarner F. Enteric flora in health and disease. Digestion. 2006;73 Suppl 1:5–12. doi: 10.1159/000089775. [DOI] [PubMed] [Google Scholar]
- 24.Cummings JH, Beatty ER, Kingman SM, Bingham SA, Englyst HN. Digestion and physiological properties of resistant starch in the human large bowel. Br J Nutr. 1996;75:733–747. doi: 10.1079/bjn19960177. [DOI] [PubMed] [Google Scholar]
- 25.Hill MJ. Intestinal flora and endogenous vitamin synthesis. Eur J Cancer Prev. 1997;6 Suppl 1:S43–S45. doi: 10.1097/00008469-199703001-00009. [DOI] [PubMed] [Google Scholar]
- 26.Gordon JI, Hooper LV, McNevin MS, Wong M, Bry L. Epithelial cell growth and differentiation. III. Promoting diversity in the intestine: conversations between the microflora, epithelium, and diffuse GALT. Am J Physiol. 1997;273:G565–G570. doi: 10.1152/ajpgi.1997.273.3.G565. [DOI] [PubMed] [Google Scholar]
- 27.Borriello SP. Bacteria and gastrointestinal secretion and motility. Scand J Gastroenterol Suppl. 1984;93:115–121. [PubMed] [Google Scholar]
- 28.Coté GA, Buchman AL. Antibiotic-associated diarrhoea. Expert Opin Drug Saf. 2006;5:361–372. doi: 10.1517/14740338.5.3.361. [DOI] [PubMed] [Google Scholar]
- 29.Bode C, Kolepke R, Schäfer K, Bode JC. Breath hydrogen excretion in patients with alcoholic liver disease--evidence of small intestinal bacterial overgrowth. Z Gastroenterol. 1993;31:3–7. [PubMed] [Google Scholar]
- 30.Addolorato G, Capristo E, Gasbarrini G, Stefanini GF. Depression, alcohol abuse and orocaecal transit time. Gut. 1997;41:417–418. doi: 10.1136/gut.41.3.417a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wegener M, Schaffstein J, Dilger U, Coenen C, Wedmann B, Schmidt G. Gastrointestinal transit of solid-liquid meal in chronic alcoholics. Dig Dis Sci. 1991;36:917–923. doi: 10.1007/BF01297141. [DOI] [PubMed] [Google Scholar]
- 32.Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res. 2009;33:1836–1846. doi: 10.1111/j.1530-0277.2009.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jokelainen K, Roine RP, Väänänen H, Färkkilä M, Salaspuro M. In vitro acetaldehyde formation by human colonic bacteria. Gut. 1994;35:1271–1274. doi: 10.1136/gut.35.9.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jokelainen K, Siitonen A, Jousimies-Somer H, Nosova T, Heine R, Salaspuro M. In vitro alcohol dehydrogenase-mediated acetaldehyde production by aerobic bacteria representing the normal colonic flora in man. Alcohol Clin Exp Res. 1996;20:967–972. doi: 10.1111/j.1530-0277.1996.tb01932.x. [DOI] [PubMed] [Google Scholar]
- 35.Visapää JP, Jokelainen K, Nosova T, Salaspuro M. Inhibition of intracolonic acetaldehyde production and alcoholic fermentation in rats by ciprofloxacin. Alcohol Clin Exp Res. 1998;22:1161–1164. [PubMed] [Google Scholar]
- 36.Ferrier L, Bérard F, Debrauwer L, Chabo C, Langella P, Buéno L, Fioramonti J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol. 2006;168:1148–1154. doi: 10.2353/ajpath.2006.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tillonen J, Väkeväinen S, Salaspuro V, Zhang Y, Rautio M, Jousimies-Somer H, Lindros K, Salaspuro M. Metronidazole increases intracolonic but not peripheral blood acetaldehyde in chronic ethanol-treated rats. Alcohol Clin Exp Res. 2000;24:570–575. [PubMed] [Google Scholar]
- 38.Atisook K, Madara JL. An oligopeptide permeates intestinal tight junctions at glucose-elicited dilatations. Implications for oligopeptide absorption. Gastroenterology. 1991;100:719–724. doi: 10.1016/0016-5085(91)80016-3. [DOI] [PubMed] [Google Scholar]
- 39.Fihn BM, Sjöqvist A, Jodal M. Permeability of the rat small intestinal epithelium along the villus-crypt axis: effects of glucose transport. Gastroenterology. 2000;119:1029–1036. doi: 10.1053/gast.2000.18148. [DOI] [PubMed] [Google Scholar]
- 40.Bjarnason I, MacPherson A, Hollander D. Intestinal permeability: an overview. Gastroenterology. 1995;108:1566–1581. doi: 10.1016/0016-5085(95)90708-4. [DOI] [PubMed] [Google Scholar]
- 41.Bungert HJ. Absorption of hemoglobin and hemoglobin iron in alcohol-induced liver injury. Digestion. 1973;9:293–308. doi: 10.1159/000197455. [DOI] [PubMed] [Google Scholar]
- 42.Worthington BS, Enwonwu C. Absorption of intact protein by colonic epithelial cells of the rat. Am J Dig Dis. 1975;20:750–763. doi: 10.1007/BF01070833. [DOI] [PubMed] [Google Scholar]
- 43.Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32:742–747. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 44.Gottfried EB, Korsten MA, Lieber CS. Alcohol-induced gastric and duodenal lesions in man. Am J Gastroenterol. 1978;70:587–592. [PubMed] [Google Scholar]
- 45.Ito S, Lacy ER. Morphology of rat gastric mucosal damage, defense, and restitution in the presence of luminal ethanol. Gastroenterology. 1985;88:250–260. doi: 10.1016/s0016-5085(85)80178-5. [DOI] [PubMed] [Google Scholar]
- 46.Victor BE, Schmidt KL, Smith GS, Miller TA. Protection against ethanol injury in the canine stomach: role of mucosal glutathione. Am J Physiol. 1991;261:G966–G973. doi: 10.1152/ajpgi.1991.261.6.G966. [DOI] [PubMed] [Google Scholar]
- 47.Persson J. Alcohol and the small intestine. Scand J Gastroenterol. 1991;26:3–15. doi: 10.3109/00365529108996478. [DOI] [PubMed] [Google Scholar]
- 48.Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am J Physiol. 1999;276:G965–G974. doi: 10.1152/ajpgi.1999.276.4.G965. [DOI] [PubMed] [Google Scholar]
- 49.Halsted CH, Robles EA, Mezey E. Distribution of ethanol in the human gastrointestinal tract. Am J Clin Nutr. 1973;26:831–834. doi: 10.1093/ajcn/26.8.831. [DOI] [PubMed] [Google Scholar]
- 50.Nosova T, Jokelainen K, Kaihovaara P, Heine R, Jousimies-Somer H, Salaspuro M. Characteristics of aldehyde dehydrogenases of certain aerobic bacteria representing human colonic flora. Alcohol Alcohol. 1998;33:273–280. doi: 10.1093/oxfordjournals.alcalc.a008391. [DOI] [PubMed] [Google Scholar]
- 51.Koivisto T, Salaspuro M. Aldehyde dehydrogenases of the rat colon: comparison with other tissues of the alimentary tract and the liver. Alcohol Clin Exp Res. 1996;20:551–555. doi: 10.1111/j.1530-0277.1996.tb01091.x. [DOI] [PubMed] [Google Scholar]
- 52.Jokelainen K, Matysiak-Budnik T, Mäkisalo H, Höckerstedt K, Salaspuro M. High intracolonic acetaldehyde values produced by a bacteriocolonic pathway for ethanol oxidation in piglets. Gut. 1996;39:100–104. doi: 10.1136/gut.39.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Basuroy S, Sheth P, Mansbach CM, Rao RK. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and L-glutamine. Am J Physiol Gastrointest Liver Physiol. 2005;289:G367–G375. doi: 10.1152/ajpgi.00464.2004. [DOI] [PubMed] [Google Scholar]
- 54.Ammori BJ, Fitzgerald P, Hawkey P, McMahon MJ. The early increase in intestinal permeability and systemic endotoxin exposure in patients with severe acute pancreatitis is not associated with systemic bacterial translocation: molecular investigation of microbial DNA in the blood. Pancreas. 2003;26:18–22. doi: 10.1097/00006676-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 55.Beger HG, Bittner R, Block S, Büchler M. Bacterial contamination of pancreatic necrosis. A prospective clinical study. Gastroenterology. 1986;91:433–438. doi: 10.1016/0016-5085(86)90579-2. [DOI] [PubMed] [Google Scholar]
- 56.Rau B, Uhl W, Buchler MW, Beger HG. Surgical treatment of infected necrosis. World J Surg. 1997;21:155–161. doi: 10.1007/s002689900208. [DOI] [PubMed] [Google Scholar]
- 57.Mier J, León EL, Castillo A, Robledo F, Blanco R. Early versus late necrosectomy in severe necrotizing pancreatitis. Am J Surg. 1997;173:71–75. doi: 10.1016/S0002-9610(96)00425-4. [DOI] [PubMed] [Google Scholar]
- 58.Sainio V, Kemppainen E, Puolakkainen P, Taavitsainen M, Kivisaari L, Valtonen V, Haapiainen R, Schröder T, Kivilaakso E. Early antibiotic treatment in acute necrotising pancreatitis. Lancet. 1995;346:663–667. doi: 10.1016/s0140-6736(95)92280-6. [DOI] [PubMed] [Google Scholar]
- 59.Exley AR, Leese T, Holliday MP, Swann RA, Cohen J. Endotoxaemia and serum tumour necrosis factor as prognostic markers in severe acute pancreatitis. Gut. 1992;33:1126–1128. doi: 10.1136/gut.33.8.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foulis AK, Murray WR, Galloway D, McCartney AC, Lang E, Veitch J, Whaley K. Endotoxaemia and complement activation in acute pancreatitis in man. Gut. 1982;23:656–661. doi: 10.1136/gut.23.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barclay GR, Scott BB, Wright IH, Rogers PN, Smith DG, Poxton IR. Changes in anti-endotoxin-IgG antibody and endotoxaemia in three cases of gram-negative septic shock. Circ Shock. 1989;29:93–106. [PubMed] [Google Scholar]
- 62.Pirisi M, Scott CA, Fabris C, Cavarape A, Federico E, Falleti E, Beltrami CA. Endotoxin priming and liver damage by experimental duodenal obstruction in the rat. Pathol Int. 2000;50:34–40. doi: 10.1046/j.1440-1827.2000.01011.x. [DOI] [PubMed] [Google Scholar]
- 63.Pastor CM, Pugin J, Kwak B, Chanson M, Mach F, Hadengue A, Frossard JL. Role of Toll-like receptor 4 on pancreatic and pulmonary injury in a mice model of acute pancreatitis associated with endotoxemia. Crit Care Med. 2004;32:1759–1763. doi: 10.1097/01.ccm.0000133020.47243.8e. [DOI] [PubMed] [Google Scholar]
- 64.Sharif R, Dawra R, Wasiluk K, Phillips P, Dudeja V, Kurt-Jones E, Finberg R, Saluja A. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut. 2009;58:813–819. doi: 10.1136/gut.2008.170423. [DOI] [PubMed] [Google Scholar]
- 65.Singh M, LaSure MM, Bockman DE. Pancreatic acinar cell function and morphology in rats chronically fed an ethanol diet. Gastroenterology. 1982;82:425–434. [PubMed] [Google Scholar]
- 66.Ammann RW, Muellhaupt B. Progression of alcoholic acute to chronic pancreatitis. Gut. 1994;35:552–556. doi: 10.1136/gut.35.4.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vonlaufen A, Phillips PA, Yang L, Xu Z, Fiala-Beer E, Zhang X, Pirola RC, Wilson JS, Apte MV. Isolation of quiescent human pancreatic stellate cells: a promising in vitro tool for studies of human pancreatic stellate cell biology. Pancreatology. 2010;10:434–443. doi: 10.1159/000260900. [DOI] [PubMed] [Google Scholar]
- 68.Lahnborg G, Friman L, Berghem L. Reticuloendothelial function in patients with alcoholic liver cirrhosis. Scand J Gastroenterol. 1981;16:481–489. doi: 10.3109/00365528109182002. [DOI] [PubMed] [Google Scholar]
- 69.Fukui H, Kitano H, Tsujii T, Morimura M, Kikuchi E, Matsumoto M, Tsujita S, Kikukawa M, Nagamoto I, Nakatani T. Effect of alcohol on the functions of Kupffer cells and splenic macrophages in rats. Alcohol Alcohol Suppl. 1993;1B:53–57. doi: 10.1093/alcalc/28.supplement_1b.53. [DOI] [PubMed] [Google Scholar]
- 70.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wyke RJ. Problems of bacterial infection in patients with liver disease. Gut. 1987;28:623–641. doi: 10.1136/gut.28.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Almdal TP, Skinhøj P. Spontaneous bacterial peritonitis in cirrhosis. Incidence, diagnosis, and prognosis. Scand J Gastroenterol. 1987;22:295–300. doi: 10.3109/00365528709078594. [DOI] [PubMed] [Google Scholar]
- 73.Madrid AM, Cumsille F, Defilippi C. Altered small bowel motility in patients with liver cirrhosis depends on severity of liver disease. Dig Dis Sci. 1997;42:738–742. doi: 10.1023/a:1018899611006. [DOI] [PubMed] [Google Scholar]
- 74.Ramachandran A, Prabhu R, Thomas S, Reddy JB, Pulimood A, Balasubramanian KA. Intestinal mucosal alterations in experimental cirrhosis in the rat: role of oxygen free radicals. Hepatology. 2002;35:622–629. doi: 10.1053/jhep.2002.31656. [DOI] [PubMed] [Google Scholar]
- 75.Schimpl G, Pesendorfer P, Steinwender G, Feierl G, Ratschek M, Höllwarth ME. Allopurinol and glutamine attenuate bacterial translocation in chronic portal hypertensive and common bile duct ligated growing rats. Gut. 1996;39:48–53. doi: 10.1136/gut.39.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spahr L, Bresson-Hadni S, Amann P, Kern I, Golaz O, Frossard JL, Hadengue A. Allopurinol, oxidative stress and intestinal permeability in patients with cirrhosis: an open-label pilot study. Liver Int. 2007;27:54–60. doi: 10.1111/j.1478-3231.2006.01382.x. [DOI] [PubMed] [Google Scholar]
- 77.Yang AL, Vadhavkar S, Singh G, Omary MB. Epidemiology of alcohol-related liver and pancreatic disease in the United States. Arch Intern Med. 2008;168:649–656. doi: 10.1001/archinte.168.6.649. [DOI] [PubMed] [Google Scholar]
- 78.Gullo L, Barbara L, Labò G. Effect of cessation of alcohol use on the course of pancreatic dysfunction in alcoholic pancreatitis. Gastroenterology. 1988;95:1063–1068. doi: 10.1016/0016-5085(88)90184-9. [DOI] [PubMed] [Google Scholar]
- 79.Lucey MR, Weinrieb RM. Alcohol and substance abuse. Semin Liver Dis. 2009;29:66–73. doi: 10.1055/s-0029-1192056. [DOI] [PubMed] [Google Scholar]
- 80.Isaji S, Suzuki M, Frey CF, Ruebner B, Carlson J. Role of bacterial infection in diet-induced acute pancreatitis in mice. Int J Pancreatol. 1992;11:49–57. doi: 10.1007/BF02925994. [DOI] [PubMed] [Google Scholar]
- 81.Foitzik T, Fernández-del Castillo C, Ferraro MJ, Mithöfer K, Rattner DW, Warshaw AL. Pathogenesis and prevention of early pancreatic infection in experimental acute necrotizing pancreatitis. Ann Surg. 1995;222:179–185. doi: 10.1097/00000658-199508000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gianotti L, Munda R, Gennari R, Pyles R, Alexander JW. Effect of different regimens of gut decontamination on bacterial translocation and mortality in experimental acute pancreatitis. Eur J Surg. 1995;161:85–92. [PubMed] [Google Scholar]
- 83.Dellinger EP, Tellado JM, Soto NE, Ashley SW, Barie PS, Dugernier T, Imrie CW, Johnson CD, Knaebel HP, Laterre PF, et al. Early antibiotic treatment for severe acute necrotizing pancreatitis: a randomized, double-blind, placebo-controlled study. Ann Surg. 2007;245:674–683. doi: 10.1097/01.sla.0000250414.09255.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boirivant M, Strober W. The mechanism of action of probiotics. Curr Opin Gastroenterol. 2007;23:679–692. doi: 10.1097/MOG.0b013e3282f0cffc. [DOI] [PubMed] [Google Scholar]
- 85.Besselink MG, van Santvoort HC, Buskens E, Boermeester MA, van Goor H, Timmerman HM, Nieuwenhuijs VB, Bollen TL, van Ramshorst B, Witteman BJ, et al. Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;371:651–659. doi: 10.1016/S0140-6736(08)60207-X. [DOI] [PubMed] [Google Scholar]
- 86.Besselink MG, van Santvoort HC, Renooij W, de Smet MB, Boermeester MA, Fischer K, Timmerman HM, Ahmed Ali U, Cirkel GA, Bollen TL, et al. Intestinal barrier dysfunction in a randomized trial of a specific probiotic composition in acute pancreatitis. Ann Surg. 2009;250:712–719. doi: 10.1097/SLA.0b013e3181bce5bd. [DOI] [PubMed] [Google Scholar]
- 87.Petrov MS, van Santvoort HC, Besselink MG, van der Heijden GJ, Windsor JA, Gooszen HG. Enteral nutrition and the risk of mortality and infectious complications in patients with severe acute pancreatitis: a meta-analysis of randomized trials. Arch Surg. 2008;143:1111–1117. doi: 10.1001/archsurg.143.11.1111. [DOI] [PubMed] [Google Scholar]
- 88.Miyasaka K, Ohta M, Takano S, Hayashi H, Higuchi S, Maruyama K, Tando Y, Nakamura T, Takata Y, Funakoshi A. Carboxylester lipase gene polymorphism as a risk of alcohol-induced pancreatitis. Pancreas. 2005;30:e87–e91. doi: 10.1097/01.mpa.0000160960.21580.ml. [DOI] [PubMed] [Google Scholar]
- 89.Rosendahl J, Witt H, Szmola R, Bhatia E, Ozsvári B, Landt O, Schulz HU, Gress TM, Pfützer R, Löhr M, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82. doi: 10.1038/ng.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]