

Abstract
Aims/Introduction
In earlier reports, we described that transgenic (Tg) mice ubiquitously expressing cryptochrome1 (CRY1) with a mutation in cysteine414 (CRY1‐AP Tg mice) show an early‐onset insulin‐secretory defect of diabetes mellitus resembling human maturity‐onset diabetes of the young (MODY). To clarify the yet undiscovered molecular pathogenesis of diabetes mellitus in which the mutant of CRY1 is involved, we examined age‐dependent characteristics of islets of CRY1‐AP Tg mice.
Materials and Methods
Immunohistochemical analyses of islets were carried out for 2‐, 4‐ and 19‐week‐old mice. Insulin contents in the pancreas and glucose‐stimulated insulin secretion of isolated islets of mice were measured at 4 weeks. Real‐time polymerase chain reaction analyses using pancreases of mice at 4 and 21 weeks‐of‐age were carried out.
Results
Already at a young stage, the proliferation of β‐cells was reduced in CRY1‐AP Tg mice. Insulin contents and the levels of glucose‐stimulated insulin secretion were lower than those of wild‐type controls in CRY1‐AP Tg mice at the young stage. The expression of insulin and glucose‐sensing genes was reduced at the young stage. At the mature stage, altered distribution and hyperplasia of α‐cells were observed in the islets of CRY1‐AP Tg mice.
Conclusions
Architectural abnormality in islets progressed with age in CRY1‐AP Tg mice. The reduced expression of insulin and glucose‐sensing genes, along with the lowered proliferation of β‐cells from an early stage, is a possible primary cause of early‐onset insulin‐secretory defect in CRY1‐AP Tg mice. Our results suggest that CRY1 is crucial for the maintenance of β‐cell function.
Keywords: Insulin secretion, Islet architecture, β‐Cell
Introduction
Maturity‐onset diabetes of the young (MODY) is a genetic subgroup of diabetes characterized by autosomal dominant inheritance and early onset, and by defective insulin secretion causing β‐cell dysfunction1. Analyses of pertinent model animals are hoped to yield information enabling the firm diagnosis of MODY and allowing prediction of its future clinical course.
Cryptochrome proteins (CRYs) play indispensable roles in the transcriptional–translational negative feedback loop of the molecular model underlying mammalian circadian rhythms, in which positive factors of the circadian locomotor output cycles kaput (CLOCK)‐brain–muscle ARNT‐like protein 1 (BMAL1) complex drive transcription of negative factors, such as Period (PER) and CRY2. Using transgenic (Tg) techniques, we previously established a mouse model of early onset insulin‐secretory defect of diabetes mellitus resembling human MODY (CRY1‐AP Tg mice), in which mutant CRY1 (cysteine414 replaced with alanine) is overexpressed ubiquitously3. The CRY1‐AP Tg mice showed not only anomalous circadian behaviors, but also symptoms of diabetes with less secretion of insulin caused by β‐cell dysfunction without accompanying insulin resistance3. One characteristic aspect of diabetes mellitus in CRY1‐AP Tg mice is that the symptoms appear from a very young age: our previous report described that Tg mice showed lower levels of serum insulin and significant glucose intolerance at 3 weeks‐of‐age4. These results suggest that the abnormality in the β‐cell functions occurs at an early stage in Tg mice. However, the molecular mechanism of the early β‐cell defect remains unknown.
At about the same time that our previous report was published4, symptoms of diabetes mellitus in Clock mutant and Bmal1 knockout mice were reported5. Those results are relevant to ours with respect to insulin‐secretory defect. Therefore, we were compelled to ascertain the detailed mechanism for CRY1‐AP Tg pathogenesis, and to compare it with that of Clock mutant and Bmal1 knockout mice for better understanding of the role of distinct clock genes in the regulation of β‐cell function.
To clarify the yet undiscovered pathogenesis of diabetes mellitus in which the mutant of clock protein CRY1 is involved, we investigated characteristic features of abnormal morphology along with molecular aspects of the pancreas and the islets in CRY1‐AP Tg mice at both the young and mature stages.
Materials and Methods
Animals
In the following, CRY1‐AP Tg mice are designated simply as Tg mice. We previously generated two transgenic lines of Tg mice: a high‐expression line (H line) and a low‐expression line (L line)3. The symptoms of diabetes in both lines are fundamentally the same, but the L line of Tg mice showed milder symptoms than mice of the H line CRY1‐AP Tg of the same age3. Therefore, H line Tg mice and their littermates (wild‐type controls) were used for experiments. Male mice were used for experiments, because Tg mouse males show symptoms of diabetes4. Mice were housed under a constant light–dark cycle (light 05.00–19.00 h). For immunohistochemical analysis of islets, pancreases were harvested at 10.00 h. For measurement of pancreatic insulin contents and quantitative real‐time polymerase chain reaction (PCR) analysis, pancreases were harvested at 17.00 h. In all experiments, animals were treated in accordance with the guidelines of Yamagata University.
Pancreatic Insulin Content, Serum Level of Insulin and Glucose‐Stimulated Insulin Secretion
Pancreatic insulin contents were measured using supernatants of acid extraction from isolated pancreases. Serum was obtained as described previously4. Pancreatic islets were isolated from mice and incubated overnight in CMRL1066 medium containing 5.6 mmol/L glucose (Sigma Chemical Co., St. Louis, MO, USA). For glucose‐stimulated insulin secretion, groups of five islets were incubated for 1 h in CMRL1066 medium containing either 5.6 mmol/L or 16.7 mmol/L glucose. The supernatants were recovered for the measurement. The insulin level was determined using an enzyme‐linked immunosorbent assay kit (Morinaga Institute of Biological Science, Inc., Kanagawa, Japan).
Immunohistochemical Analysis and Morphometry of Islets
We carried out immunohistochemical analyses for insulin, glucagon, pancreatic and duodenal homeobox‐1 (PDX‐1), and V‐maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA). Immunostaining of paraffin‐embedded pancreas sections (3 μm) using rabbit anti‐insulin antibody (1:2000 H‐86; Santa Cruz Biotechnology Inc., Dallas, TX, USA), rabbit anti‐glucagon antibody (1:100 Ab‐1; Thermo Fisher Scientific Inc., Waltham, MA, USA), rabbit anti‐MafA (1:100 NBP1‐00121; Novus Biologicals Inc., Littleton, CO, USA) and rabbit anti‐PDX‐1 (1:250 KR059; TransGenic Inc., Kumamoto, Japan) was carried out using a kit (Histofine Simple Stain MAX‐PO; Nichirei Biosciences Inc., Tokyo, Japan). For detection of MafA and PDX‐1, the mounted sections were heated at 120°C for 5 min in citrate buffer (pH 6.0) by autoclave heating for antigen retrieval before being incubated with the antibodies. The sections were counterstained with hematoxylin for identification of pancreatic islets. For islet size measurements, islets were displayed on a computer monitor through a microscope connected to a charge‐coupled device camera (Leica Camera AG, Solms, Germany). For each experimental group, 4‐week‐old and 19‐week‐old mice were used. Measurements and calculations were carried out with sections that had been prepared from four to five pancreases for each experimental group (three sections per mouse). For each section, both the whole pancreas and the islet area were traced manually. The relative value of the islet area was calculated as the ratio of the total area of the islet to that of the pancreas. The measurements of α‐cell and β‐cell areas were carried out fundamentally as described in an earlier report4. Approximately 30 islets were measured per mouse. The relevant dimensions were analyzed using software (Adobe Photoshop; Adobe Systems Inc., San Jose, CA, USA). The total β‐cell mass and α‐cell mass were calculated from the data of the pancreas weight, islet area and β‐ or α‐cell area. For measurement of the proliferation of β‐cells, 2‐week‐old mice were used. After autoclave heating, pancreas sections were labeled successively with guinea pig anti‐insulin (1:100 PA1‐26938; Thermo Scientific), Alexa Fluor 488 goat anti‐guinea pig immunoglobulin G (IgG) conjugate (Molecular Probes Inc., Eugene, OR, USA), rabbit anti‐proliferating cell nuclear antigen (PCNA; 1:400 FL‐261; Santa Cruz Biotechnology Inc.) and Alexa Fluor 594 goat anti‐rabbit IgG conjugate (Molecular Probes Inc.). Nuclear DNA was counterstained with 4,6‐diamidino‐2‐phenylindole (DAPI; 0.5 μg/mL; Wako Pure Chemical Industries Ltd., Osaka, Japan). Images of islets (approximately 30 islets per mouse) were captured by the CCD camera and analyzed using software (Doctor file viewer; Finggal Link Co. Ltd., Tokyo, Japan) for β‐cell proliferation and mean fluorescence intensity of staining for insulin.
Quantification of Messenger Ribonucleic Acid by Real‐Time PCR
Total ribonucleic acid (RNA) was extracted from frozen pancreas using isogen reagent (Nippon Gene Co. Ltd., Toyama, Japan). Complementary deoxyribonucleic acids (cDNA) were synthesized from RNA using a reverse transcription kit (QuantiTect; Qiagen Inc., Venlo, the Netherlands). Real‐time PCR analysis was carried out using SsoFast EvaGreen Supermix (Bio‐Rad Laboratories Inc., Hercules, CA, USA) with the CFX96 Real‐Time System (Bio‐Rad Laboratories Inc.) according to the protocols provided by the manufacturer. The housekeeping gene, hypoxanthine‐guanine phosphoribosyltransferase (HPRT), was used as the endogenous control for quantification. Results obtained from analyses using CFX Manager (version 1.6; Bio‐Rad Laboratories Inc.) are expressed as relative values with respect to HPRT levels using the ΔΔCT method. Relevant primer sets are shown in Appendix S1.
Statistical Analysis
For each experiment, t‐tests were used to compare the mean values for Tg mice with those of wild‐type controls. Differences between means were inferred as significant for P < 0.05. Data are presented as means ± standard error.
Results
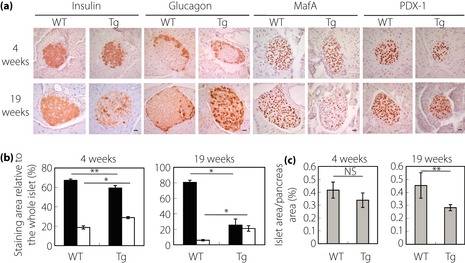
β‐Cell Area, α‐Cell Area, and Islet Size in Tg Mice
To examine how characteristic aspects of the islets change with age, we carried out analyses of the young 2–5‐week‐old mice as described here. Then we compared the results with those obtained for mature 19–21‐week‐old mice. We first carried out immunohistochemical analysis for insulin. At 4 weeks, the amount of the β‐cell area of Tg mice was slightly smaller (by 11.7% in the ratio) than that of wild‐type controls without showing any anomalous appearance (Figure 1a,b). In contrast, the area of β‐cells in Tg mice was much smaller than that of wild‐type controls at 19 weeks‐of‐age (Figure 1a,b), reinforcing the results presented in our earlier report4. We then carried out immunohistochemical analyses for glucagon. At 4 weeks‐of‐age, although no gross abnormality in the distribution of α‐cells was observed in Tg mice (Figure 1a), the area of α‐cells was slightly larger than that of wild‐type controls (Figure 1b). In stark contrast, at 19 weeks‐of‐age, the area of α‐cells per islet was clearly higher in Tg mice than in wild‐type controls (Figure 1a,b). Furthermore, α‐cells were distributed abnormally in Tg mice. The α‐cells in Tg mice were observable throughout the islet (Figure 1a), whereas in wild‐type mice, α‐cells were distributed mainly in the peripheral region of the islets (Figure 1a). Our results show that the loss of β‐cell area and abnormality in the islet architecture progress with age in Tg mice. We subsequently carried out immunohistochemical analyses for transcription factors MafA and PDX‐1. Regarding MafA and PDX‐1 at 4 weeks‐of‐age, no evident change in islet staining was observed (Figure 1a). At 19 weeks‐of‐age, MafA‐positive nuclei were far fewer in Tg mice (Figure 1a). For PDX‐1, the positive nuclei were fewer than in wild‐type controls (Figure 1a). To ascertain whether the alterations in the islets of Tg mice at the mature stages described here are associated with the change in the islet size, we examined the percentage of the islet area relative to the whole pancreas area (Figure 1c). At 4 weeks‐of‐age, no significant difference was found between the islet areas of Tg mice and wild‐type controls, which suggest that the development of islets in Tg mice is almost normal (Figure 1c). At 19 weeks‐of‐age, the islet area in Tg mice was significantly smaller than in wild‐type controls (Figure 1c). We measured bodyweight, pancreas weight, and blood glucose level at both 4 and 19 weeks‐of‐age (Figure S1). At 4 weeks, Tg mice showed slightly lower bodyweight than that of wild‐type controls (Figure S1). This result confirms our previous results showing mild retardation in growth at the young stage4. Regarding the pancreas weight, no significant difference was found between Tg mice and wild‐type controls at either 4 or 19 weeks‐of‐age (Figure S1). Consequently, it was estimated that the total β‐cell mass was reduced by approximately 12% in Tg mice compared with that of wild‐type controls at 4 weeks. In addition, the estimated total α‐cell mass in Tg mice was approximately 2.2‐fold as large as that in wild‐type controls at 19 weeks. No significant difference was found between the levels of blood glucose in Tg mice and wild‐type controls at 4 weeks, but at the mature stage Tg mice showed a higher level of blood glucose (Figure S1), also confirming the results presented in our earlier report4.
Figure 1.
Immunohistochemical analyses of pancreatic islets and pancreatic islet areas. (a) Immunostaining of pancreas sections of wild‐type (WT) and transgenic mice ubiquitously expressing cryptochrome1 with a mutation in cysteine414 (Tg) at 4 weeks (upper panels) and 19 weeks (lower panels) for insulin, glucagon, V‐maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA) and pancreatic and duodenal homeobox‐1 (PDX‐1). Bar, 20 μm. (b) Quantitative analyses of the insulin‐stained and glucagon‐stained area in the islet of 4‐week‐old (left panel) and 19‐week‐old (right panel) mice. Approximately 30 islets were measured per mouse. Data are means ± standard error for five mice per group (solid bars, insulin; open bars, glucagon; **P < 0.05, *P < 0.01, t‐test). (c) Relative pancreatic islet areas in 4‐week‐old (left panel) and 19‐week‐old (right panel) mice. Data are expressed as a percentage relative to the whole pancreas area. Data are means ± standard error (Tg mice: 4 weeks, n = 4; 19 weeks, n = 4; wild‐type controls: 4 weeks, n = 4; 19 weeks, n = 5; **P < 0.05, t‐test).
Expression of mRNAs in the pancreas of Tg mice
To analyze the molecular mechanism of the pathogenesis further, real‐time PCR analyses were carried out using pancreatic cDNA derived from young and mature mice (Figure 2). We used total pancreatic cDNA for real‐time PCR analyses. The expression levels of transcription factors, PDX‐1, neurogenic differentiation 1 (NeuroD1) and hepatocyte nuclear factor‐1α (Hnf‐1α) were examined in Tg mice pancreases at both 4 and 21 weeks‐of‐age (Figure 2). The results show that no significant difference in the expression levels was observed at 4 weeks‐of‐age, but the mRNA levels of NeuroD1, Hnf‐1α and PDX‐1 were significantly lower than those of wild‐type mice at 21 weeks‐of‐age (Figure 2). We quantified the respective mRNA levels of insulin1, insulin2 and glucose‐sensing genes (GLUT2 and glucokinase; Figure 2). The expression levels of these genes were lower in Tg mice than in wild‐type controls already by 4 weeks‐of‐age (Figure 2). The differences in the amount of these mRNAs in Tg mice compared with those of wild‐type controls were greater at 21 weeks‐of‐age (Figure 2). To evaluate the circadian clock function in the pancreas of Tg mice, we quantified mRNA levels of the genes associated with the circadian clock function: period1, period2 and rev‐erbα, and D‐element binding protein (DBP)2. The mRNA levels of these genes were markedly lower in both the young and mature stages in Tg mice than in wild‐type controls (Figure 2), indicating disruption of the circadian clock function in the pancreases of Tg mice from young stages.
Figure 2.
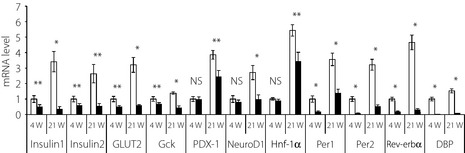
Messenger ribonucleic acid (mRNA) expression of insulin (insulin1, insulin2) and glucose‐sensing genes [GLUT2, glucokinase (Gck)], transcription factor genes [pancreatic and duodenal homeobox‐1 (PDX‐1), neurogenic differentiation 1 (NeuroD1) and hepatocyte nuclear factor‐1α (Hnf‐1α)] and clock genes [period1, period2, rev‐erbα, D‐element binding protein (DBP)] in the pancreas at 4 weeks‐of age (4 w) and 21 weeks‐of‐age (21 w). In each representation, the value for the wild‐type controls at 4 weeks was set to 1. Data are means ± standard error (open bars, wild‐type controls: 4 weeks, n = 8; 21 weeks, n = 5; solid bars, Tg mice: 4 weeks, n = 7; 21 weeks, n = 4; **P < 0.05, *P < 0.01, t‐test).
Insulin Contents, Serum Insulin Levels and Glucose‐Stimulated Insulin Release in the Islet
We compared pancreatic insulin contents of Tg mice with those of wild‐type controls at 4 and 5 weeks‐of‐age (Figure 3a). Insulin contents in Tg mice were significantly lower than those in respective wild‐type controls. Serum insulin levels were significantly reduced in Tg mice at 4 weeks‐of‐age compared with wild‐type controls (Figure 3b). The levels were reduced considerably in Tg mice compared with wild‐type controls at 19 weeks‐of‐age (Figure 3b). Additionally, we measured the capability of insulin release using the islets derived from mice at 4 weeks. Glucose (16.7 mmol/L)‐stimulated insulin release of islets from Tg mice was significantly lower than those of wild‐type controls (Figure 3c). At the control condition (5.6 mmol/L glucose), no significant difference was found between Tg mice and wild‐type controls (Figure 3c).
Figure 3.
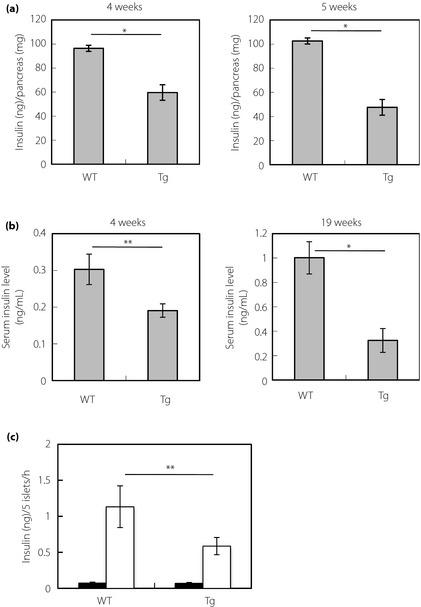
Insulin contents and serum insulin levels of mice fed ad libitum, and glucose‐stimulated insulin release in the islet. (a) Insulin contents in the pancreas at 4 weeks (left panel) and 5 weeks (right panel). Data are means ± standard error for seven mice (4 weeks) or six mice (5 weeks) per group (*P < 0.01, t‐test). (b) Serum insulin levels at 4 weeks (left panel) and 19 weeks [right panel; wild‐type controls (WT): 4 weeks, n = 11; 19 weeks, n = 4; transgenic mice ubiquitously expressing cryptochrome1 with a mutation in cysteine414 (Tg): 4 weeks, n = 11; 19 weeks, n = 5; **P < 0.05, *P < 0.01, t‐test]. (c) Insulin secretion from isolated pancreatic islets of mice at 4 weeks. Insulin secretion was measured at 5.6 mmol/L (solid bars) and 16.7 mmol/L glucose (open bars; wild‐type controls, n = 6; Tg mice, n = 7; **P < 0.05, t‐test).
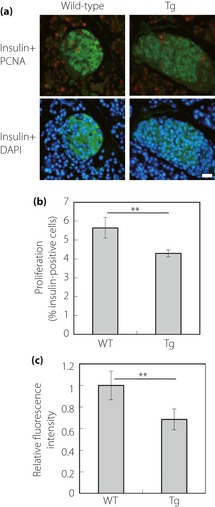
Proliferation of β‐Cells in Tg Mice
To investigate the possible mechanisms of the lowered β‐cell area in Tg mice compared with wild‐type controls (Figure 1a,b), we carried out terminal deoxynucleotidyl transferase‐mediated dUTP‐biotin nick end labeling analysis of pancreas sections in Tg mice at 4 and 19 weeks‐of‐age. The results showed no sign of increased apoptosis in the islet in either the young or mature stage compared with respective wild‐type controls (Figure S2). Then we carried out immunohistochemical analysis of β‐cell proliferation using pancreas sections from Tg mice and wild‐type controls at 2 weeks‐of‐age by staining for the cell proliferation marker PCNA and insulin with DAPI nuclear counterstaining (Figure 4). The percentage of β‐cells in islets that were positive for PCNA was lower in pancreas sections from Tg mice than in those from wild‐type controls (Figure 4b). Consequently, it seems likely that the lowered β‐cell proliferation contributes mainly to the age‐dependent loss of β‐cell area in Tg mice. We quantified the fluorescence intensity of staining for insulin in each islet of the 2‐week‐old mice (Figure 4c). Staining intensities in Tg mice were lower than those in wild‐type controls (Figure 4c).
Figure 4.
β‐Cell proliferation and insulin expression in pancreatic islets at 2 weeks‐of‐age. (a) Co‐immunostaining for insulin and proliferating cell nuclear antigen (PCNA). Pancreas sections from wild‐type (left panel) and Tg (right panel) mice were co‐stained with antibodies to insulin (green) and PCNA (red), and counterstained with 4,6‐diamidino‐2‐phenylindole (DAPI; blue) for nuclear staining. Bar, 20 μm. (b) Quantification of β‐cell proliferation. The cells that were positive for both PCNA and insulin were quantified as a percentage of the total number of insulin‐positive cells. Data are means ± standard error for five mice per group (**P < 0.05, t‐test). (c) Relative fluorescence intensity of staining for insulin in islets. The value for the wild‐type controls was set to 1. Data are means ± standard error for five mice per group (**P < 0.05, t‐test).
Discussion
The expression from the transgene in the islet was shown in mice harboring the transgene with the same promoter as that we used for the present study7. Therefore, the present results (Figure 4) suggest that the mutant CRY1 expressed in the islet depresses the proliferation of β‐cells from the young stage in Tg mice. Recent studies have shown that insulin plays an important role in β‐cell proliferation8. The loss of β‐cells itself might further depress the proliferation rate of the remaining β‐cells in Tg mice. Reportedly, interorgan communication through autonomic nerves can regulate β‐cell proliferation9. Furthermore, the function of CRY plays some role in the regulation of the autonomic nerve activity10. Therefore, the disturbance of neuronal signals from outside the pancreas might contribute to the lowered proliferation of β‐cells in Tg mice.
Transcription factors PDX‐1, NeuroD1, Hnf‐1α and MafA, which are expressed in pancreatic β‐cells, are known to be involved in regulation of the transcription of insulin11. In addition to that for insulin, these transcription factors regulate the transcription of various genes that are important for β‐cell function. For instance, Hnf‐1α13, MafA14 and PDX‐117 regulate the expression of GLUT2. PDX‐1 also regulates the transcription of glucokinase18. Specific mutations in the genes of Pdx‐1, NeuroD1 and Hnf‐1α are associated with MODY1. The respective expressions of PDX‐1, NeuroD1, Hnf‐1α and MafA were lower in the pancreas of Tg mice at the mature stage (Figures 1 and 2), which conforms with the lower expressions of insulin, glucokinase and GLUT2 at the mature stage (Figure 2). Chronic hyperglycemia results in failure in the function of β‐cells, including decreased expression of insulin, which is known as glucose toxicity19. Reported results showed that during chronic hyperglycemia the expression of MafA is reduced, and decreased expression of MafA is implicated in the reduction of insulin expression20. Furthermore, in MafA‐deficient mice, the expression of PDX‐1 and NeuroD1 was reportedly reduced in islets, in conjunction with the decrease of insulin and GLUT216. In MafA‐deficient mice, age‐dependent abnormal islet architecture has also been reported16. We reported previously that the higher level of blood glucose than that of wild‐type controls became apparent from 6 weeks‐of‐age in Tg mice, and that the level of blood glucose increases in an age‐dependent manner4. From the present study, we confirmed that no significant difference exists in the level of blood glucose between Tg mice and wild‐type controls at 4 weeks (Figure S1). Therefore, at the mature stage (19–21 weeks‐of‐age), the islets in Tg mice are under chronic high‐glucose conditions, although there can be little influence of glucose toxicity on Tg mice in the young stage before 6 weeks‐of‐age (2–5 weeks‐of‐age). Consequently, the reduced expression of MafA as a result of chronic hyperglycemia might contribute to the reduction of the transcription factors, insulin and glucose‐sensing genes, and to the development of the abnormality in the islet in Tg mice at the mature stage (Figures 1 and 2). On the contrary, the observed glucose intolerance4, the reduction of insulin contents and serum insulin levels (Figure 3 and ref 4), as well as the reduction of insulin secretion (Figure 3) in Tg mice at 3 and 4 week, cannot be caused by the side‐effect of glucose toxicity. Also the reduction of mRNA levels of insulin and glucose‐sensing genes at 4 weeks (Figure 2) cannot be due to the side‐effect of glucose toxicity.
Reportedly, Clock mutant and Bmal1 global/pancreas‐specific knockout mice show impaired insulin release and diabetes5. Importantly, results of physiological experiments using isolated islets from Clock mutant and Bmal1 global/pancreas‐specific knockout mice have shown that the impaired insulin release in the mice is attributable to malfunction in the latest stage of stimulus–secretion coupling5. Along with the result, it was also shown that no substantial reduction occurs in insulin contents of the isolated islets or the pancreas in Clock mutant mice5, or in pancreas‐specific Bmal1 knockouts6. In contrast, in Tg mice, the mRNA level of insulin (Figure 2) as well as the insulin contents (Figure 3) were reduced to approximately 50–60% in the pancreas compared with wild‐type controls already at the young stage (4–5 weeks‐of‐age). These results show that the pathogenesis of diabetes in Tg mice is not identical to that in Clock mutant and Bmal1 knockout mice.
We previously showed that the L line of Tg mice, which do not show drastic circadian abnormalities (only a slightly longer free‐running period3), display diabetes similarly to the H line of Tg mice3. These results suggest that the effect of abnormalities of circadian behavior is not the main cause of the diabetes of Tg mice, although the secondary effect of abnormal circadian activity cannot be ruled out entirely.
In addition to that of insulin, the level of GLUT2 mRNA was 49% lower in Tg mice than in wild‐type controls at 4 weeks (Figure 2). Furthermore, the level of glucokinase mRNA was 32% lower in Tg mice than in wild‐type controls at 4 weeks (Figure 2). As stated previously, β‐cell mass was lower by approximately 12% in Tg mice than in wild‐type controls at 4 weeks. The differences in quantities of these mRNAs in Tg mice and in wild‐type controls (Figure 2) were greater than the loss of β‐cell mass, suggesting that the expression of insulin, glucokinase and GLUT2 genes in each β‐cell is reduced in Tg mice already at the young stage. The level of insulin protein per islet was indeed lower than that of wild‐type controls at 2 weeks (Figure 4c). In addition, the level of GLUT2 protein per islet was evidently lower than that of wild‐type controls at 4 weeks (Figure S3). Heterozygous mutations in glucokinase cause the pathogenesis of MODY21. Reportedly, a heterozygous mutation of the glucokinase gene, either globally21 or in the pancreatic β‐cells22, results in impaired insulin secretion to glucose and hence diabetes similar to human MODY. Glut2‐deficient mice show a diminished glucose‐induced insulin secretion and symptoms of diabetes23. Based on the present results and those in the literature, simultaneous reduction in the expression of insulin, glucokinase and GLUT2 can account for the reduced level of serum insulin (Figure 3b and ref 4) along with decreased glucose‐stimulated insulin release (Figure 3c) with significant glucose intolerance in Tg mice at the young stage4. At 4 weeks, mRNAs of insulin, glucokinase and GLUT2 genes were reduced, but no discernible reduction of the expression of their regulators, PDX‐1, NeuroD1, Hnf‐1α and MafA, was observed (Figure 1 and 2). These results suggest that the mutant CRY1 directly affects the transcription of insulin, glucokinase and GLUT2 genes without affecting the aforementioned transcription factors. Results showed that, among clock‐related genes, the expression of DBP was markedly depressed in Tg mice (Figure 2). Reportedly, DBP is highly expressed in islets in humans24. Furthermore, its potential importance in the transcriptional regulation of β‐cell expressing genes including GLUT2 was also shown24. Consequently, the depression of DBP expression by mutant CRY1 might be responsible for the reduced expression of insulin, glucokinase and GLUT2 in Tg mice.
In summary, the present results strongly suggest that the reduced expression of insulin, glucokinase and GLUT2 genes, and the lowered proliferation of β‐cells from the early stages are possible causes of diabetes in Tg mice. Our findings also suggest that CRY1 plays some important part in the maintenance of pancreatic β‐cell function. These results might provide new insight into the pathophysiology of non‐obese early‐onset diabetes and provide a basis for the development of new therapeutic agents.
Supplementary Material
Appendix S1 | Primer sets used in real‐time polymerase chain reaction analysis.
Figure S1 | Blood glucose level, bodyweight, and pancreas weight of wild‐type controls and Tg mice at 4 and 19 weeks‐of‐age.
Figure S2 | Apoptosis of wild‐type controls and Tg mice at 4 and 19 weeks‐of‐age.
Figure S3 | Immunostaining for GLUT2 of mice at 4 weeks‐of‐age.
Acknowledgements
The authors thank Professor S Yagihashi and Dr H Mizukami of Hirosaki University for kind technical advice related to the islet separation. This study was supported in part by Grants‐in‐Aid from the (Japanese) Ministry of Education, Culture, Sports, Science, and Technology (21590429, 24590473). The authors have declared that no conflicts of interest exist.
(J Diabetes Invest, doi: 10.1111/jdi.12080, 2013)
References
- 1.Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity‐onset diabetes of the young. N Engl J Med 2001; 345: 971–980 [DOI] [PubMed] [Google Scholar]
- 2.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature 2002; 418: 935–941 [DOI] [PubMed] [Google Scholar]
- 3.Okano S, Akashi M, Hayasaka K, et al Unusual circadian locomotor activity and pathophysiology in mutant CRY1 transgenic mice. Neurosci Lett 2009; 451: 246–251 [DOI] [PubMed] [Google Scholar]
- 4.Okano S, Hayasaka K, Igarashi M, et al Non‐obese early onset diabetes mellitus in mutant cryptochrome1 transgenic mice. Eur J Clin Invest 2010; 40: 1011–1017 [DOI] [PubMed] [Google Scholar]
- 5.Marcheva B, Ramsey KM, Buhr ED, et al Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010; 466: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadacca LA, Lamia KA, deLemos AS, et al An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia 2011; 54: 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawamoto S, Niwa H, Tashiro F, et al A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre‐mediated recombination. FEBS Lett 2000; 470: 263–268 [DOI] [PubMed] [Google Scholar]
- 8.Johnson JD, Alejandro EU. Control of pancreatic beta‐cell fate by insulin signaling: the sweet spot hypothesis. Cell Cycle 2008; 7: 1343–1347 [DOI] [PubMed] [Google Scholar]
- 9.Imai J, Katagiri H, Yamada T, et al Regulation of pancreatic β cell mass by neuronal signals from the liver. Science 2008; 322: 1250–1254 [DOI] [PubMed] [Google Scholar]
- 10.Tanida M, Yamatodani A, Niijima A, et al Autonomic and cardiovascular responses to scent stimulation are altered in cry KO mice. Neurosci Lett 2007; 413: 177–182 [DOI] [PubMed] [Google Scholar]
- 11.Andrali SS, Sampley ML, Vanderford NL, et al Glucose regulation of insulin gene expression in pancreatic beta‐cells. Biochem J 2008; 415: 1–10 [DOI] [PubMed] [Google Scholar]
- 12.Emens LA, Landers DW, Moss LG. Hepatocyte nuclear factor 1 alpha is expressed in a hamster insulinoma line and transactivates the rat insulin I gene. Proc Natl Acad Sci USA 1992; 89: 7300–7304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamagata K, Nammo T, Moriwaki M, et al Overexpression of dominant‐negative mutant hepatocyte nuclear fctor‐1 alpha in pancreatic beta‐cells causes abnormal islet architecture with decreased expression of E‐cadherin, reduced beta‐cell proliferation, and diabetes. Diabetes 2002; 51: 114–123 [DOI] [PubMed] [Google Scholar]
- 14.Matsuoka TA, Kaneto H, Stein R, et al MafA regulates expression of genes important to islet beta‐cell function. Mol Endocrinol 2007; 21: 2764–2774 [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Brun T, Kataoka K, et al MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007; 50: 348–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang C, Moriguchi T, Kajihara M, et al MafA is a key regulator of glucose‐stimulated insulin secretion. Mol Cell Biol 2005; 25: 4969–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waeber G, Thompson N, Nicod P, et al Transcriptional activation of the GLUT2 gene by the IPF‐1/STF‐1/IDX‐1 homeobox factor. Mol Endocrinol 1996; 10: 1327–1334 [DOI] [PubMed] [Google Scholar]
- 18.Watada H, Kajimoto Y, Miyagawa J, et al PDX‐1 induces insulin and glucokinase gene expressions in alphaTC1 clone 6 cells in the presence of betacellulin. Diabetes 1996; 45: 1826–1831 [DOI] [PubMed] [Google Scholar]
- 19.Robertson RP, Harmon J, Tran PO, et al Glucose toxicity in β‐cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003; 52: 581–587 [DOI] [PubMed] [Google Scholar]
- 20.Matsuoka TA, Kaneto H, Miyatsuka T, et al Regulation of MafA expression in pancreatic beta‐cells in db/db mice with diabetes. Diabetes 2010; 59: 1709–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grupe A, Hultgren B, Ryan A, et al Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell 1995; 83: 69–78 [DOI] [PubMed] [Google Scholar]
- 22.Terauchi Y, Sakura H, Yasuda K, et al Pancreatic beta‐cell‐specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J Biol Chem 1995; 270: 30253–30256 [DOI] [PubMed] [Google Scholar]
- 23.Guillam MT, Hümmler E, Schaerer E, et al Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut‐2. Nat Genet 1997; 17: 327–330 [DOI] [PubMed] [Google Scholar]
- 24.Allaman‐Pillet N, Roduit R, Oberson A, et al Circadian regulation of islet genes involved in insulin production and secretion. Mol Cell Endocrinol 2004; 226: 59–66 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 | Primer sets used in real‐time polymerase chain reaction analysis.
Figure S1 | Blood glucose level, bodyweight, and pancreas weight of wild‐type controls and Tg mice at 4 and 19 weeks‐of‐age.
Figure S2 | Apoptosis of wild‐type controls and Tg mice at 4 and 19 weeks‐of‐age.
Figure S3 | Immunostaining for GLUT2 of mice at 4 weeks‐of‐age.