

Insulin resistance, a state of reduced responsiveness to insulin, is associated with obesity and is the major pathogenic indicator of metabolic syndrome. Although major advances have been made in unraveling the underlying defects that cause insulin resistance, many of the pathways and regulators that connect insulin to its downstream metabolic effects are not completely understood. Epidemiological studies have demonstrated that excess lipid accumulation in non‐adipose tissues, such as liver and skeletal muscle, promotes a lipotoxic state and results in insulin resistance, highlighting the importance of lipid accumulation in the pathogenesis of metabolic syndrome.
In normal physiology, feeding triggers the release of insulin, which suppresses glucose production and activates lipogenesis in the liver; in obesity and type 2 diabetes, insulin fails to suppress glucose production and lipogenesis is paradoxically enhanced. At the molecular level, increased lipogenesis observed in the insulin‐resistant state is due, at least in part, to dysregulation of the master transcriptional regulator of lipogenesis, namely sterol regulatory element‐binding protein (SREBP)‐1c. The SREBP family, originally identified as basic helix‐loop‐helix (bHLH) leucine zipper transcription factors by Brown and Goldstein1, is involved in the regulation of the entire complement of genes necessary for the synthesis of fatty acids (FAs), triglycerides (TGs), and cholesterol. The SREBP family consists of three isoforms: SREBP‐1a, SREBP‐1c, and SREBP‐2. The biosynthesis of FAs and TGs is controlled by SREBP‐1c, the upregulation of genes involved in cholesterol metabolism is preferentially governed by SREBP‐2, and SREBP‐1a activates both FA and cholesterol biosynthetic pathways. Expression of SREBP‐2 is induced under conditions of sterol depletion, whereas SREBP‐1c expression is under the control of insulin, glucose, and FAs. Insulin activates SREBP‐1c through at least two mechanisms: (i) it increases SREBP‐1c transcription; (ii) it increases the proteolytic cleavage of SREBP‐1c from an inactive endoplasmic reticulum (ER) membrane‐bound precursor to release the N‐terminal domain, which is capable of translocating to the nucleus to activate transcription (Figure 1).
Figure 1.
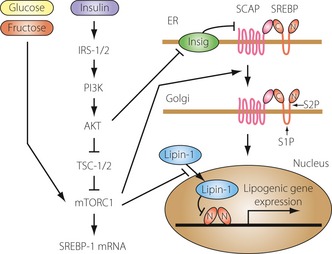
Insulin‐dependent and ‐independent signaling pathways involved in the regulation of sterol regulatory element‐binding protein (SREBP)‐1c. There are multiple signals that regulate expression, endoplasmic reticulum (ER)‐to‐Golgi transport, and the proteolytic cleavage of SREBP‐1c. Insulin‐dependent and ‐independent pathways are able to activate mammalian target of rapamycin complex 1 (mTORC1) and increase SREBP‐1c mRNA. Insulin‐dependent pathways are required for the processing of SREBP‐1c and maximal lipogenic gene expression. C, SREBP C‐terminal fragment; Insig, insulin‐induced gene; IRS, insulin receptor substrate; N, SREBP N‐terminal fragment; PI3K, phospha‐tidylinositol 3‐kinase; S1P, site 1 protease; S2P, site 2 protease; SCAP, SREBP cleavage‐activating protein; TSC, tuberous sclerosis complex.
In hepatocytes, insulin signaling through phosphatidylinositol 3‐kinase (PI3K) and AKT results in activation of SREBP‐1c expression and accumulation of nuclear SREBP‐1c protein; the major downstream effector of PI3K/AKT is mammalian target of rapamycin complex 1 (mTORC1)2. This conclusion is based on studies showing that insulin‐stimulated SREBP activation and lipogenesis are both blocked by the mTORC1 inhibitor rapamycin. Activation of PI3K/AKT leads to direct phosphorylation of tuberous sclerosis complex (TSC) 1/2 and proline‐rich Akt substrate 40 kDa (PRAS40), resulting in mTORC1 activation. In TSC1/2‐null mouse embryonic fibroblasts (MEFs), mTORC1 is constitutively active and independent of upstream signals. SREBP‐1 is activated in TSC1/2 null MEFs and SREBP‐1 activation is inhibited following rapamycin treatment, indicating that mTORC1 activation is sufficient to stimulate SREBP‐1 activity. Inhibition of S6 kinase, a downstream target of mTORC1, inhibits SREBP‐1 processing in TSC1/2‐null MEFs, but fails to block insulin‐induced SREBP‐1c activation in primary rat hepatocytes, suggesting the existence of S6 kinase‐independent SREBP‐1c regulation by mTORC1.
A recent study revealed an additional important component of the AKT regulatory pathway, lipin‐1. Lipin‐1, a phosphatidic acid phosphatase (PAP) and transcriptional coactivator, is a direct substrate of mTORC1 and regulator of nuclear SREBP activity3. Lipin‐1 phosphorylation by mTORC1 blocks its nuclear localization and activates both SREBP‐1 and SREBP‐2. Conversely, expression of a non‐phosphorylated form of lipin‐1 results in nuclear localization of lipin‐1, reduced nuclear SREBP levels, and altered SREBP localization to the nuclear periphery. Knockdown of lipin‐1 in liver‐specific Raptor‐knockout mice restores SREBP‐1 activation. These studies implicate lipin‐1 as an insulin‐dependent downstream effector of mTORC1 that regulates SREBP activity. Although the inhibitory effect requires the intact PAP enzyme active site, the exact mechanism involved remains unknown. Interestingly, SREBP‐1 directly activates lipin‐1 transcription in human hepatoma cells, providing a possible mechanism for negative feedback regulation of SREBP activity.
Insulin regulates SREBP activity through additional mechanisms. Inhibition of PI3K by chemical inhibitors or expression of dominant‐negative AKT inhibits SREBP ER‐to‐Golgi transport and proteolytic activation. Consistent with this, insulin stimulates ER‐to‐Golgi transport of SREBP‐1c by promoting its phosphorylation and association with coat protein complex II (COPII) vesicles. In mouse liver, transport of SREBP is also regulated by AKT signaling through the control of insulininduced gene‐2 (Insig‐2) levels. Two differently regulated mRNA transcripts (Insig‐2a and Insig‐2b) code for Insig‐2, although Insig‐2a is the predominant hepatic transcript. Insulin negatively regulates Insig‐2a mRNA expression in an AKTdependent manner, resulting in a decreased Insig protein pool and increased ER‐to‐Golgi transport of SREBP. Overall, insulin acts at multiple regulatory steps to control SREBP activity.
Lipogenesis is an insulin‐sensitive pathway that is overactivated in the liver of insulin‐resistant obese subjects; however, the mechanisms involved in this pathway remain a matter of debate. There are two possible explanations for the increase in lipogenesis observed in the insulin‐resistant state. The first possibility is that insulin, despite its ability to control glucose homeostasis, retains its ability to stimulate lipogenesis. As suggested by Brown and Goldstein1, the signaling pathways used by insulin to stimulate SREBP‐1c could remain intact in obesity and type 2 diabetes, even as the pathways that regulate glucose metabolism become resistant. The resulting hyperinsulinemia, acting through insulin‐sensitive signaling pathways, would drive SREBP‐1c and lipogenesis to excess. Activation of lipogenesis through the unfolded protein response (UPR) of ER and SREBP‐1c stimulation is certainly an interesting possibility, because it explains, at least in part, the paradox of having to admit that one side of the insulin signaling pathway is still sensitive whereas the other is resistant.
Alternatively, it is possible that, in the insulin‐resistant state, lipogenesis is driven by insulin‐independent pathways. Particular attention has recently been focused on dietary carbohydrates, because excessive consumption of carbohydrates, particularly in the form of sweetened beverages, has risen in parallel with the prevalence of obesity, diabetes, and fatty liver disease. However, it has been difficult to discern whether carbohydrates increase lipogenesis directly or indirectly through stimulation of insulin secretion. Nonetheless, the fact that carbohydrate loads in humans can induce hypertriglyceridemia even in the presence of somatostatin, which inhibits insulin secretion, indicates that at least some of the carbohydrates may be metabolized independent of insulin signaling. Consistent with this theory, we have shown that SREBP‐1c can be induced by carbohydrate feeding even in streptozotocin‐treated mice, which are insulin deficient4. Furthermore, a recent study using liver‐specific insulin receptor‐knockout (LIRKO) mice showed that, in the physiological context of feeding, hepatic insulin signaling is not required for SREBP‐1c induction5. Feeding induces SREBP‐1c mRNA in LIRKO livers, although not to the extent observed in controls. A high fructose diet also induces SREBP‐1c and lipogenic gene expression in LIRKO livers. Insulin signaling becomes more important in the physiological context of obesity, because knockdown of the insulin receptor in ob/ob mice using antisense oligonucleotides abolishes the induction of SREBP‐1c and its targets by obesity and ameliorates steatosis. This, together with data in mice with liver‐specific knockout of both insulin receptor substrate (IRS)‐1 and IRS‐2, or AKT2, suggests that nutritional regulation of SREBP‐1c and lipogenic genes may be completely independent of insulin as long as sufficient carbohydrates are available, but the further induction by obesity and type 2 diabetes is entirely insulindependent (Figure 1).
In human type 2 diabetes, hyperinsulinemia, dietary carbohydrates, and other lipogenic stimuli are present. Recent studies suggest that identifying and targeting the specific signaling pathways by which insulin stimulates lipogenesis, as well as decreasing dietary carbohydrates, could be extremely effective in reducing SREBP‐1c levels and ameliorating insulin resistance and hepatic steatosis in type 2 diabetes. Further unveiling of the molecular mechanisms that regulate SREBP‐1c gene expression and activity in obesity and diabetes will hopefully result in effective ways to overcome insulin resistance and provide a basis for beneficial treatment of type 2 diabetes.
References
- 1.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell 1997; 89: 331–340 [DOI] [PubMed] [Google Scholar]
- 2.Bakan I, Laplante M. Connecting mTORC1 signaling to SREBP‐1 activation. Curr Opin Lipidol 2012; 23: 226–234 [DOI] [PubMed] [Google Scholar]
- 3.Peterson TR, Sengupta SS, Harris TE, et al mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011; 146: 408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsuzaka T, Shimano H, Yahagi N, et al Insulin‐independent induction of sterol regulatory element‐binding protein‐1c expression in the livers of streptozotocin‐treated mice. Diabetes 2004; 53: 560–569 [DOI] [PubMed] [Google Scholar]
- 5.Haas JT, Miao J, Chanda D, et al Hepatic insulin signaling is required for obesity‐dependent expression of SREBP‐1c mRNA but not for feeding‐dependent expression. Cell Metab 2012; 15: 873–884 [DOI] [PMC free article] [PubMed] [Google Scholar]