

Abstract
Objectives
To elucidate the possible ways by which hydroxyurea molecules affect globin chain (α or β-like) synthesis.
Methods
A total of 23 thalassemia intermedia patients (13 male and 10 female) aged between 5 and 26 years were treated for five months with 15 mg/(kg·day) of hydroxyurea. Hemoglobins electrophoresis and globin chain electrophoresis was performed on each sample at different time points before and during the treatment.
Results
Fetal hemoglobin increased significantly in most patients and average episodes of transfusion decreased. Both Gγ and Aγ-globin chains increased significantly and α-globin:Nonα-globin chain as well as Gγ-globin:Aγ globin chains ratios decreased.
Conclusions
Improvement in α:non-α ratio and consequent decrease of free α-globin chain might be the cause of beneficial effects of hydroxyurea therapy. Two patients who felt better didn't show significant increase in their fetal hemoglobin level, and this is in contradiction with the hypothesis claiming that the HbF level increase is the cause of such therapeutic effect. In spite of the unclear mechanism of action of this drug, hydroxyurea therapy had noticeable impacts on thalassemia intermedia and also sickle cell disease and even patients suffering from thalassemia major.
Keywords: Thalassemia intermedia, Globin chain, AUT-PAGE, Hydroxyurea, Hemoglobin
1. Introduction
Hemoglobin (Hb) is the iron-containing metalloprotein in the red blood cells (RBCs) of all vertebrates. It is responsible for delivering oxygen to tissues and in human, it has many variants. In a normal subject, according to the stage of development such as embryonic, fetal and adult, hemoglobins are present. Hemoglobin's quaternary structure comes from a roughly tetrahedral arrangement of its four subunits. The most common hemoglobin type is a tetramer consisting of two α-like (ζ or α) and two non-α-like globin chain (ε,Gγ,Aγ,δ or β) subunits non-covalently bounded, each made of 141 and 146 amino acid residues, respectively. Globin gene activation is coordinated during development. The composition of the four globin chains determines the hemoglobin type. For example: HbA (α2β2), HbA2 (α2δ2), fetal hemoglobin (HbF) (α2γ2) mutations in the globin genes cause hemoglobinopathies that are inherited diseases[1].
Thalassemia has an autosomal recessive pattern of inheritance and is a hemoglobinopathy along with hemolytic anemia which is determined by hypochromic, microcytic and decreased RBCs lifetime caused by reduced or absence of one or more globin chain synthesis. The fundamental abnormality in thalassemia is impaired production resulting in an imbalanced ratio of either α or non-α globin chains. According to the chain involved, the two major thalassemia categories are α-thalassemia (reduced α globin chain synthesis) and β-thalassemia (reduced beta globin chain synthesis).
β-thalassemia intermedia (TI), the milder form of disease is a term used to define a group of patients with β-thalassemia, in whom the clinical severity of the disease is ranging between the mild symptoms of the β-thalassemia trait and the severe manifestations of β-thalassemia major. Clinical diagnosis of this disease is based on the hemoglobin concentration 6-7 g/dL at the time of diagnosis and in many cases regular blood transfusions are not necessary[2]. Despite tremendous advances in diagnosis of the genetic and molecular basis of this syndrome in the past decade, the diagnosis and classification of diseases is still largely dependent on clinical syndromes. In practical terms, a child who is referred to the clinic at the first time with hemoglobin levels less than 7 g/dL at the age of two or more years, with or without splenomegaly, is categorized as thalassemia intermedia[3].
New estimations suggest 300 000 annual births of hemoglobinopathies, among which sickle cell disease (SCD) and the thalassemia represent the majority of cases[4],[5]. In Iran with an approximately 15 000 thalassemic patients[1],[6]; the most common single gene disorder is β-thalassemia. In north of Iran, near the Caspian sea, and south close to the Persian Gulf the prevalence of β-thalassemia is about 10% and in other parts of the country about 4%-8% prevalence is expected[6],[7]. Patients with β-thalassemia who have high levels of HbF, especially patients suffering hereditary persistence of fetal hemoglobin, have fewer problems to the extent that they may be transfusion independent and they may live almost normally[8]–[10]. Three classes of HbF-inducing agents have been investigated[11] for the treatment of β-thalassemia including chemotherapeutics [for example: hydroxyurea (HU)[12]–[15], 5-azacytidine[13],[14], decitabine[16], vinblastin[17], thalidomide[18]], pervanadate compounds[19], short-chain fatty acid derivatives (for example: sodium phenylbutyrate, arginine butyrate, hydroxycarbamide[20] and Isobutyramide sodium 2,2- dimethylbutyrate[16],[18], recombinant human erythropoietin[16],[21]), and recently some inducers from the natural world (for example: angelicin and linear psoralens, resveratrol, rapamycin[22] have also been reported to be efficient in increasing HbF levels[23]). New oral agents that stimulate both fetal globin gene expression and erythropoiesis are also promising candidates for HbF induction[21].
HU was shown to reduce the rate of painful crises in adult patients who have at least three painful crises per year and have possible beneficial effects on HbF concentration increase in patients with SCD. In spite of the controversy over the exact mechanism of induction of HbF by 5-azacytidine and HU, considerable progress has been made in the development of these agents as drugs can induce HbF in patients with hemoglobin disorders[16].
The aim of this study was to elucidate the possible ways by which HU molecules affect globin gene expression and which of the globin chains α or β-like is augmented in the presence of this drug. The determination of the location and the mechanism of action of this medication and similar drugs will determine the study direction of more efficient drugs or a multi-drug combination. There are only a few studies on the effect of this medication on α:non-α and Gγ:Aγ ratio[24]. Thus, analyzing these indices in TI patients seemed necessary in order to better elucidate the mechanism of action of HU on β globin gene cluster expression.
2. Materials and methods
2.1. Patients
This cross sectional clinical trial study, the same as a lot of other similar studies, has experienced the problems that led to restrictions on the sample size such as: medical ethics, patient consent, having inclusion criteria and lack of exclusion criteria. Inclusion criteria can be described as: 1) A person who suffers thalassemia intermedia; 2) Not suffering from another disease same as viral disease and don't need drug therapy; 3) Don't take interferon; 4) Has the ability to see the doctor several times; 5) With the doctor's diagnose, be able for sampling and HU therapy. And exclusion criteria was: 1) Decreasing a large number of platelets (thrombocytopenia); 2) Significant decrease in WBCs (leukocytopenia); 3) Dissatisfaction of patient or his family.
A total of 26 thalassemia intermedia subjects aged between 5 and 26 years attending thalassemia department of the Imam Reza hospital on the north of Iran were initially included in this study. The average starting age of transfusion was 3.3 years. None of them had viral disease nor needed drug therapy or took interferon. All patients or their parents consented to participate to this study. Three of them were excluded from the study after the first month of treatment due to thrombocytopenia and consequently the number of cases became 23 (13 male and 10 female). All remaining patients were treated for five months with 15 mg/(kg·day) of HU. About 6 mL blood was withdrawn from each patient before, and 2.5 months and 5 months after starting the HU therapy. About 14 patients were spelenectomized prior to this study.
2.2. Cellular and biochemical analyses
Complete blood count (CBC), reticulocyte count, HbF concentration measurement, hemoglobins electrophoresis, acid acetic-urea-triton X100- poly acrylamide gel electrophoresis (AUT-PAGE) was performed on each sample. CBC was done by automated hematology analyzer Sysmex K-1000 Howell Jully body instrument (Sysmex, USA). Packed cells were prepared from blood samples collected in EDTA, by using isotonic normal saline (0.9%) and were used for estimating Hb amount by means of acetate cellulose according to the separation instruction from Helena company. After electrophoresis, plates were stained using ponceau S and destained three times with 5% acetic acid solution. Then, dehydration was done in the presence of absolute methanol, and finally the slides were cleared by means of clear aid followed by heating at 80 °C in the oven. The slides were scanned with a densitometer at 540 nm wavelength and the acquired curves were analyzed by appropriate software. In this method (pH=8.6) the movement of Hemoglobins from negative to positive (cathode to anode) are HbA, HbF and HbA2 respectively.
HbF was measured by resistance to alkali denaturation method of Singer[25]. Pure water (hypotonic solution) was used for RBCs lysis, then the solution was saturated by mean of ammonium sulphate and the pH was changed by using NaOH. All Hbs except HbF, salted out. Optical density of filtered solution containing HbF was measured at 540 nm. Globin chains were separated by AUT-PAGE by a modification of Rovera's method which consists of a high decrease in electrophoresis time.
Acrylamide gels with 0.67% bisacrylamide were prepared. Preelectrophoresis was performed first on acetic acid buffer at 200 V for 45 min and then at the presence of 2-mercaptoethanol at 150 V for 30 min. Samples were prepared in acetic acid and 2-mercaptoethanol and electrophoresed for 4.5 h at 150 V[23]. After electrophoresis, the gels were stained in coomassie blue for 24 h and then destained for few days. All instruments were calibrated by standard methods.
2.3. Statistical analysis
In order to statistical analysis, student's t-test by SPSS software was used and the sign test were used for nonparametric methods[24].
3. Results
HU therapy caused dramatic changes in the average episodes of transfusion from (10±7.7) per 5 months before treatment to (3.6±3.8) per 5 months during treatment and all patients were satisfied and felt better. In clinical history, all patients reported an increase in their ability for physical and intellectual activities and due to the decrease of transfusion, less need to desferal therapy. Table 1 shows the blood parameters before and during treatment at 2.5 and 5 months. Blood cells indices not only can be a response of the medication efficiency, but also a symbol of patient's healthcare. Variations in CBC before, 2.5 and 5 months after treatment are shown in Table 1.
Table 1. Blood parameters before and during treatment at 2.5 and 5 months.
| Blood indices | Onset | After 2.5 months | After 5 months | P value |
| WBC | 499.00±17.80 | 566.00±20.40 | 747.00±35.10 | >0.05 |
| RBC | 3.19±0.46 | 3.32±0.43 | 3.58±0.40 | <0.05 |
| HCT | 24.99±2.52 | 25.86±2.26 | 2.83±2.48 | <0.05 |
| MCV | 76.90±6.08 | 77.93±6.24 | 80.79±6.36 | <0.05 |
| MCH | 23.60±1.93 | 24.06±2.17 | 25.06±2.07 | <0.05 |
| MCHC | 29.56±2.44 | 31.10±1.65 | 31.62±1.74 | <0.05 |
| Platelet | 520.52±214 | 480.57±187.00 | 405.22±188.00 | <0.05 |
| Retic count | 8.90±3.14 | 6.55±3.42 | 4.71±2.17 | <0.05 |
In order to evaluate whole Hb molecules and look for Hb variants, the study took advantage of Hb electrophoresis on acetate cellulose. Figure 1 shows a slide containing 8 samples of acetate cellulose electrophoresis, and an example of scanning curve may be seen in Figure 2.
Figure 1. A Helena acetate cellulose electrophoresis slide with 8 samples.
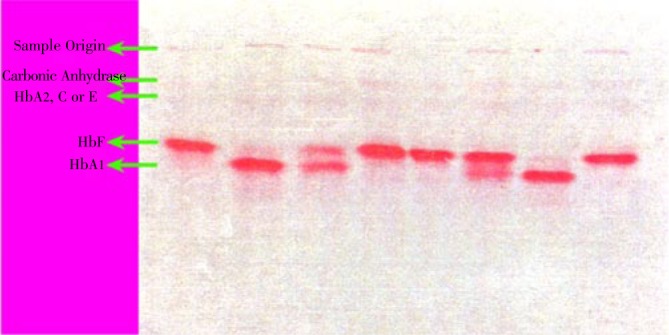
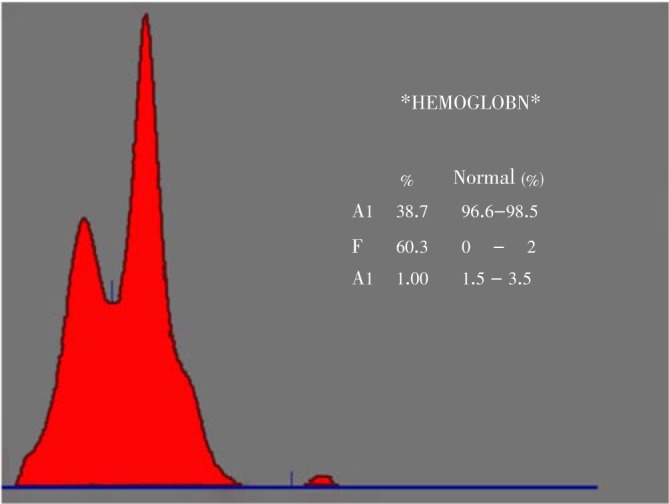
Figure 2. A scanned curve of Helena acetate cellulose electrophoresis slide for 1 sample.
Then globin chains were measured separately by AUT-PAGE. Figure 3 represents the electrophoresis pattern. The bands absorbance on the gel was measured at 590 nm by densitometry.
Figure 3. Tubes with poly acrylamide gels after staining and destaining.
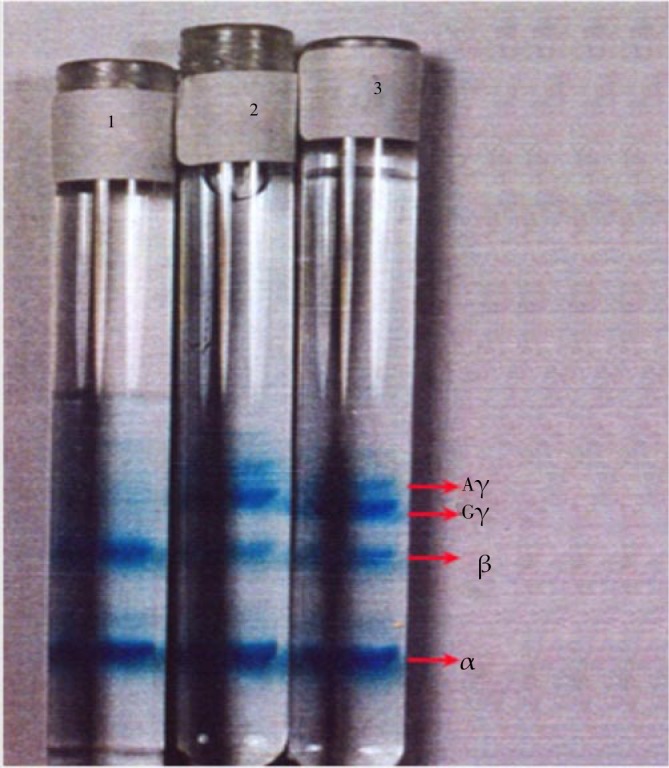
The increment of γ chains after 2.5 and 5 months is so clear. From the top to the bottom, bands correspond to carbonic anhydrase and globin chains Aγ, Gγ, β and α respectively.
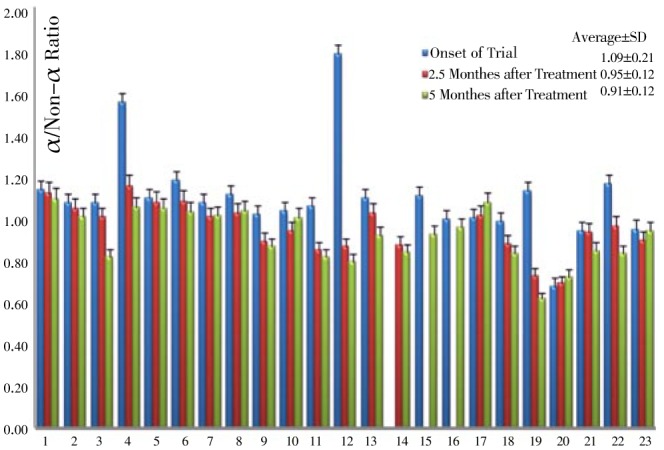
Figure 4 represents a density pattern obtained for one of the samples. Total hemoglobins, HbF, Gγ:Aγ as well as α:non-α chains ratio were assessed before, 2.5 and 5 months after HU therapy. Table 2 represents those molecular indices. Variations of HbF as well as α/non-α chains for all 23 patients are represented in Figure 5 and 6 respectively.
Figure 4. A scanned graph of one sample of polyacrylamide gel electrophoresis.

Table 2. Molecular parameters before and during treatment at 2.5 and 5 months.
| Parameters | Onset | After 2.5 months | After 5- months | P value |
| Total Hb | 7.64±0.91 | 7.99±0.85 | 8.74±0.98 | <0.05 |
| HbF | 6.90±4.77 | 11.71±5.69 | 15.48±6.09 | <0.05 |
| Gγ | 13.19±7.67 | 15.20±10.42 | 1 6.27±10.98 | <0.05 |
| Aγ | 5.78±4.46 | 8.49±5.28 | 11.52±6.45 | <0.05 |
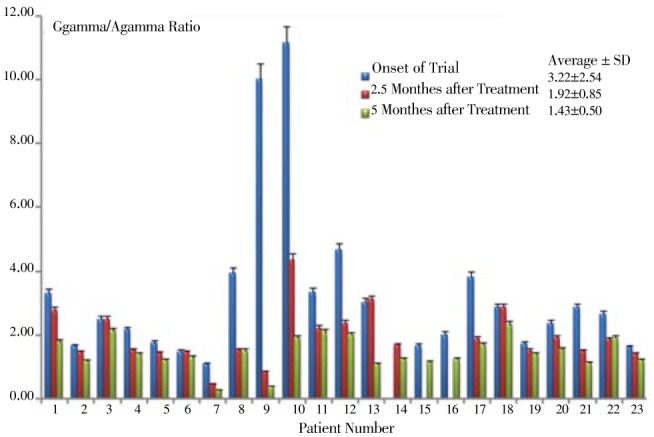
| Gγ/Aγ | 3.22±2.54 | 1.92±0.85 | 1.43±0.50 | <0.05 |
| α/non-α | 1.09±0.21 | 0.95±0.12 | 0.91±0.12 | <0.05 |
Figure 5. Varriation of HbF at onset, 2.5 and 5 months of clinical trial by alkali denaturation test.

Figure 6. Varriation of α/non-α chains ratio at onset, 2.5 and 5 months of clinical trial.
In patients number 9 and 18 a slight decrease in the whole process of treatment and in patient number 6, a slight decrease in the second phase followed by a slight increase in the 3rd phase of sampling was observed in Figure 5. In the other 20 patients HbF increased significantly (P<0.05).
According to Table 2, it seems that the amount of α chain decreased gradually and shifted to make a balance with non-α. Also the amount of β chain decreased from (29.27±13.28) percent at the treatment onset to (24.72±17.08) at the end of the treatment. Therefore, the ratio of α:non-α decreased from (1.09±0.21) to (0.91±0.12) (Figure 6).
Gγ variation was discordant in this survey as it increased in some patients and decreased in others. But according to Table 2, it's average amount increased during the treatment period, from (13.19±7.67) at the beginning of treatment to (16.27±10.98) at the end of this study, which shows a significant change. (P<0.05). Although Aγ showed an alternative increase and decrease during the study, but as shown in Table 2, an overall increase in the amount of Aγ is observed from (5.78±4.46) at the beginning of treatment to (11.52±6.45) at the end, which is considered as a significant increase(P<0.05).
Gγ:Aγ showed a decrease in almost all patients during the period of drug administration (Table 2 Figure 7); because of an excess increase in two patients at the first stage of the treatment, the average of this ratio shows a slight increase, despite a moderate decrease in other patients. By discarding the data corresponding to the above cited two patients, an important decrease in the ratio of Gγ:Aγ from (2.22±2.54)% to (1.43±0.50)% at the end of the treatment period can be noticed.
Figure 7. Varriation of Gγ/Aγ chains ratio at onset, 2.5 and 5 months of clinical trial.
In AUT-PAGE method for α, β, Gγ and Aγ measurement in three patients, A sample of three samples lost and the data are average of two stages sampling.
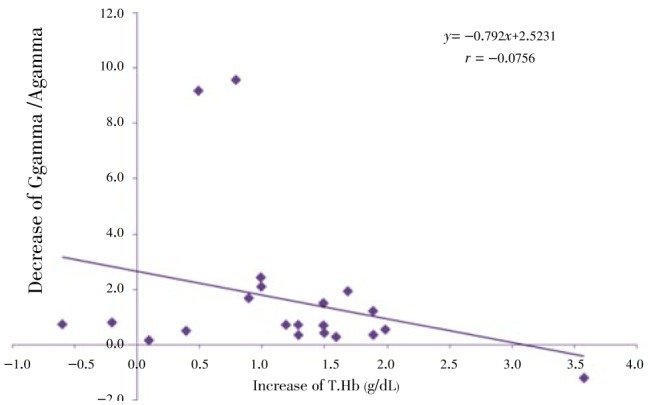
Regression analysis of some indices is presented in Figure 8–12. The correlation between Gγ and HbF which is represented in Figure 8 shows fewer changes. As shown in Figure 9, the correlation between HbF and total Hb during the treatment is r1=0.29, r2=0.23, r3=0.25 respectively which shows a decrease of the correlation between these two indices. Although there is a direct relationship between total Hb and HbF increase, because of a higher increase of HbF in HU treated patients, the correlation between these two indices decreased slightly during treatment. The correlation between Aγ and HbF is represented in Figure 10. The correlation between total Hb increase with α:non-α decrease and Gγ:Aγ ratios is represented in Figure 8 and 10. According to Figure 11 a weak correlation exists between total Hb and Gγ:Aγ ratio. The comparison of Figure 11 with Figure 12 which represents the correlation of total HB with α:non-α, suggests that the correlation between these two latter indices (r=0.20) is more than the regression coefficient of total Hb with Gγ:Aγ (r=0.07).
Figure 8. Correlation between Gγ and HbF at onset, 2.5 and 5 months of clinical trial.
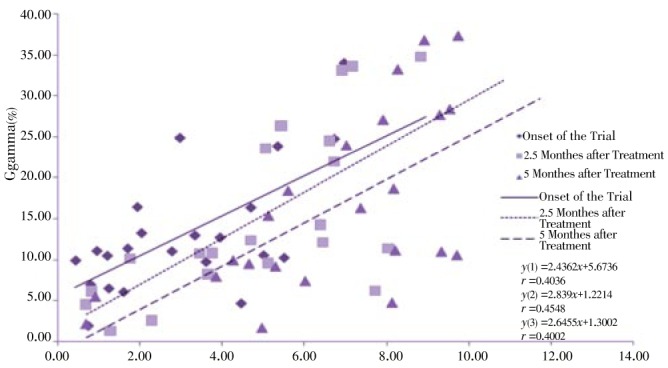
Figure 12: Correlation between decrease of α/Non-α and increase of T.Hb at onset, 2.5 and 5 months of clinical trial.
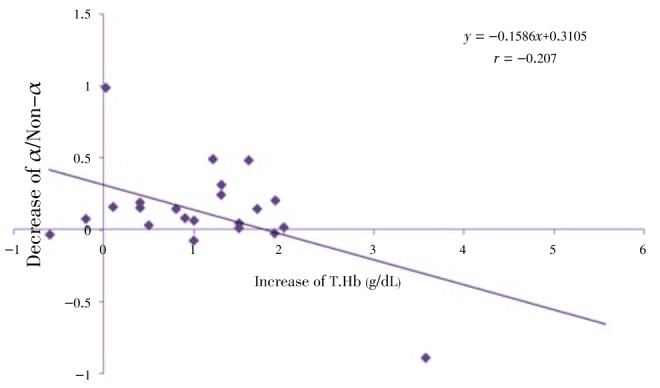
Figure 9. Correlation between HbF and total Hb at onset, 2.5 and 5 months of clinical trial.

Figure 10. Correlation between Aγ and HbF at onset, 2.5 and 5 months of clinical trial.
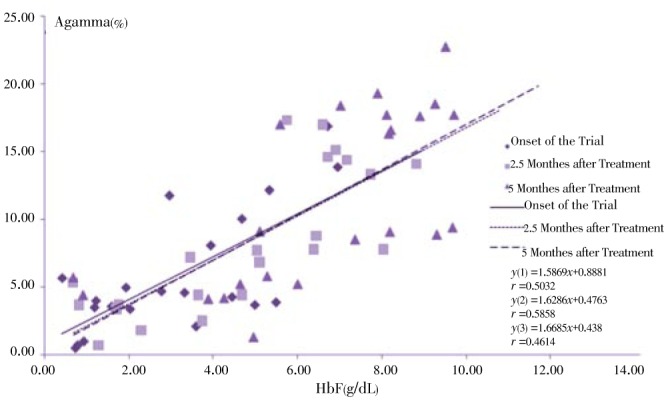
Figure 11. Correlation between decrease of Gγ/Aγ and increase of T.Hb at onset, 2.5 and 5 months of clinical trial.
4. Discussion
Many studies demonstrate an increase of HbF in SCD patients after HU administration[26],[27]. However investigations on the effect of this drug on thalassemia had different and sometimes contradictory results, showing an increase of β and γ chains in some studies or little effect in others[28]–[31]. In this study some beneficial effects of HU administration due to an increase of the γ globin chains was observed. Beneficial effects were also suggested by other studies on Sickle/βThal, Hb lepore/ βthal, βThal/HbE and other TI patients.
The amount of WBC indices in this study during the 3 phases of sampling showed no significant change in the patients (P>0.05). Although WBC is not an important criteria in TI therapy but because of its role in health and survival, it can may be considered as another exclusion criteria during HU therapy.
An increase in the amount of RBCs which are the only carriers of hemoglobins in blood circulation and are considered as the most important problem in thalassemia patients, was observed in this study (P<0.05) which may have caused a significant decrease in the transfusion episodes. Contrary to some reports[32]–[34], in this study, we found a significant increase in the amount of total Hb (P<0.05) which is in agreement with the findings of Kosaryan et al[35]. An increase in vital ability, physical activities and a decrease of transfusion dependence and also a lower bone deformities in patients treated with HU can also be attributed to the increase of total Hb. HCT increase reflects RBC and MCV increase and consequently is an expected result. Significant increase of total Hb and HCT similar to those observed in our study, was also previously reported[35]–[37] although Karimi et al. did not observe such changes[38].
Other indices of blood cells like MCV and MCH also had a significant increase which is in agreement with other studies[39],[40]. An increase in the amount of HCT, MCV and MCH is due to natural erythropoiesis improvement. Like the study of Dixit et al[41], the results of this study have shown a significant increase in MCHC whereas some drugs have shown different effects in thalassemic patients[42]. Although we noticed a significant decrease in the average level of patients platelet, but this decrial was not so important to necessitate a treatment arrest. A significant decrease in reticulocytes count level which was also reported by Dixit et al[41], might reflect bone marrow stress associated with hypoxia (anoxia) improvement, although it can also be due to the toxicity of the drug on bone marrow.
However, the fact that the increase in Hb decreases the number of reticulocytes and supports the erythropoiesis improvement hypothesis. Most of the latest studies performed on the effect of HU on TI are in agreement with the results obtained from this study showing an increase in the level of HbF[21],[43]. In fact, the first observed hematologic response of patients to treatment is an increase of HbF level. Zeng and his colleagues believed that HU was acting by increasing the production of β globin chain[44] but we observed an increase in γ chain and HbF consequently. Studies performed on k562 cell lines reported an increase in the amount of γ chains[18]. Thus, the increase in the production of HbF might have a relation with the improvement of α/non-α after HU therapy.
In severe β-globin disorders, bone marrow failure resulting in aplastic crises (secondary to myelotoxicity) may be followed by the temporary reduction in the reticulocytes count, LDH, bilirubin and a temporary increase of the HbF level. Such effects are mostly followed by a higher decrease in the amount of reticulocytes counts (up to zero) and continuous decrease in the hemoglobin level, while the average level of the total red blood cells will remain unchanged.
In the present study, blood globin chains were separated and the α:non-α and Gγ:Aγ ratio were surveyed. According to our results, a short decrease in the amount of α globin which might reflect an increase in γ chains occurred. The amount of β globin also decreased slightly and in a similar manner, this decrease might not correspond to the absolute amount of β chains but also reflects the increase of the total level of globin chains. The amount of Gγ and Aγ increased dispersedly in the patients and the average level of both chains showed a significant increase (P<0.05). However, the total amount of gamma chains increase in patients is more important[14],[45]. Zeng and his colleagues suggested that the increase of total Hb is due to the increase of β globin level not γ chain[46]. Most of the studies performed to date are in agreement with our results concerning the improvement in α:non-α ratio being the cause of beneficial effects of HU therapy and the decrease of free α-globin chain[47]. A significant decrease in the above ratio was also reported by Fucharoen and his colleagues who also reported a decrease in Gγ:Aγ while this decrease was not significant[48].
The reason of observed effects in HU therapy in TI patients has not been clearly demonstrated. Studies on k562 cell lines show that the increase of Aγ and Gγ chains at cellular level is in accordance with the effect of this drug at the genomic expression level. Researches on DNA methylation, showed that unlike 5-azacytidine, the mechanism of HU action is not related to the methylation modifications or -158 XmnI SNP[49] and probably mediated by additional mechanisms[50]. Furthermore, two patients who feel better did not show significant increase in their HbF level, which is in contradiction with the hypothesis claiming that the HbF level increase is the cause of such therapeutic effect. In spite of the unclear mechanism of action of this drug, HU therapy had noticeable impacts on TI and also SCD and even patients suffering from thalassemia major[51] and its limited side effects[28] make it a good candidate for these diseases therapy.
The comparison of two regression analysis (Gγ and Aγ compared with HbF) shows that the increase in HbF level is more correlated with Aγ augmentation. This idea is further supported by the decrease of Gγ:Aγ ratio and increased correlation between Aγ and HbF during HU therapy as well as the increase of the average of the two latter indices[11]. Finally we deduced that the imposed effect of HU on Aγ is more than Gγ.
The advantages of using this medication appears to be more than an increase in HbF levels, but the increase of the α:non-α ratio can be considered as one of the main reasons. Since, disproportion in α:non-α ratio have been known as a molecular agent of problems observed in thalassemia in many studies, Thus the imbalance between two groups of globin molecules causes accumulation of α-like globin chains and especially α-globin chain in patients with β thalassemia. This condition provides insoluble deposits inside the red blood cells that causes deformation, instability and destruction of the cell membrane (decreased cellular deformability). Although the increment of HbF and HbA2 is a pleasant event in patients with β-thalassemia but because of high affinity for oxygen and more stability of oxygen binding that mentioned for two molecules in comparison to HbA1, the efficiency of the HbF and HbA2 in compared with HbA1 is reduced. Our observations of the 23 patients indicate that satisfaction and positive changes created in daily life of above patients are beyond of the significant changes observed in HbF, γ-globin chains, Gγ/Aγ ratio, α:non-α ratio, retic count, etc. This results is confirmed based on the life quality improvement in 3 patients that have been observed any increase in the level their HbF. Therefore, it appears that supplementary reasons should be found in association with the benefits of this drug in patients with thalassemia such as the enhancement in production nitric oxide, structural changes in the binding and release of oxygen from hemoglobin, improvement in tissues oxygen consumption, expression of some genes unrelated to the globin chains, change in level of some hormones, etc.
Acknowledgments
This research was supported by Iran University of Medical Sciences and Health Services, Mazandaran University of medical sciences and Health Services and North Research Center-Pasteur Institute of Iran, (Grant No:791203). The authors gratefully acknowledge Mrs. Sara Ghobadi Rad and Mrs. Effat Hayati from North research Center-Pasteur Institute of Iran, Vesal Center of Iranian Blood Transfusion Organization and Imam Reza Hospital of Mazandaran University of Medical Sciences and Health Services for providing some facilities necessary to carry out our research work.
Comments
Background
Increased level of HbF may ameliorate the clinical crisis of β-thalassemia and SCD. Many agents stimulate HbF augmentation in vivo such as 5-azacytidine, butyrate, and HU. The mechanisms of action of these agents are different and not yet understood clearly. So in order to design new drugs, understanding the mechanism of these drugs at the molecular level can be helpful to modern advances.
Research frontiers
The present study demonstrates HU beneficial effects on the patient condition by increasing HbF. Also, α-like:β-like globin chain ratio, Gγ and Aγ has been studied to better understand the biochemical and genetic effects of HU.
Related reports
Recent studies showed that HU and its derivatives can stimulate the in vivo erythropoiesis and fetal globin production in thalassemia patients, a substantial and persistent increase in total hemoglobin level and a significant decrease in blood transfusion. Increased HbF production in β-thalassemia/HbE patients, with an improvement in the α:non-α globin ratios and probably, the effectiveness of erythropoiesis, can be achieved using HU.
Innovations and breakthroughs
Although HU is prescribed to thalassemia patients as appropriate and FDA-approved medication, this research study introduced new insights in globin chain production and molecular mechanisms of this drug. AUT-PAGE method with an improvement on electrophoresis duration is another novelty of this research.
Applications
Improvement of electrophoretic dissociation method of metalloproteins can be used in research and diagnostic laboratories and augmentation of HbF.
Peer review
Over all the paper is very informative and gives scientific information. This is an interesting study where the authors have studied the influence of HU on WBC, total Hb, HbF, globin chains synthesis and ratio, α:non-α and Gγ:Aγ. HU showed effective influence on clinical condition of thalassemia intermedia patients. This study is important and useful to those who work on thalassemia intermedia, drug inducers of HbF, electrophoresis, gene therapy etc.
Footnotes
Foundation Project: Supported by Iran University of Medical Sciences and Health Services, Mazandaran University of medical sciences and Health Services and North Research Center-Pasteur Institute of Iran, (Grant No:791203).
Conflict of interest statement: We declare that we have no conflict of interest.
References
- 1.Haghshenas M, Zamani J. Thalassemia. Shiraz: Shiraz University of Medical Sciences Publishing Center; 1997. [Google Scholar]
- 2.Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. doi: 10.1186/1750-1172-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cappellini MD, Musallam KM, Taher AT. Insight onto the pathophysiology and clinical complications of thalassemia intermedia. Hemoglobin. 2009;33(S1):S145–S159. doi: 10.3109/03630260903351528. [DOI] [PubMed] [Google Scholar]
- 4.Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;26(S1):S3–S6. doi: 10.1016/S0268-960X(12)70003-6. [DOI] [PubMed] [Google Scholar]
- 5.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331–4336. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soteh Hashemi MB, Akhavan Niaki H, Kowsarian M, Aliasgharian A, Banihashemi A. Frequency of beta-globin gene mutations in beta-thalassemia patients from east of Mazandaran. J Mazandaran Univ Med Sci. 2008;18(67):17–25. [Google Scholar]
- 7.Habibzadeh F, Yadollahie M, Merat A, Haghshenas M. Thalassemia in Iran; an overview. Arch Iran Med. 1998;1:27–34. [Google Scholar]
- 8.Amato A, Cappabianca MP, Perri M, Zaghis I, Grisanti P, Ponzini D, et al. Interpreting elevated fetal hemoglobin in pathology and health at the basic laboratory level: new and known γ- gene mutations associated with hereditary persistence of fetal hemoglobin. Int J Lab Hematol. 2013 doi: 10.1111/ijlh.12094. [DOI] [PubMed] [Google Scholar]
- 9.Zhang C, Wu ZK. Molecular pharmacological basis of the YiSui ShenXu Granule in β-thalassemia therapy. J Ethnopharmacol. 2008;120(3):437–441. doi: 10.1016/j.jep.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 10.Mabaera R, West RJ, Conine SJ, Macari ER, Boyd CD, Engman CA, et al. A cell stress signaling model of fetal hemoglobin induction: what doesn't kill red blood cells may make them stronger. Exp Hematol. 2008;36(9):1057–1072. doi: 10.1016/j.exphem.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 11.Boosalis MS, Castaneda SA, Trudel M, Mabaera R, White GL, Lowrey CH, et al. Novel therapeutic candidates, identified by molecular modeling, induce γ-globin gene expression in vivo. Blood Cells Mol Dis. 2011;47(2):107–116. doi: 10.1016/j.bcmd.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karimi M. Hydroxyurea in the management of thalassemia intermedia. Hemoglobin. 2009;33(S1):S177–S182. doi: 10.3109/03630260903351809. [DOI] [PubMed] [Google Scholar]
- 13.Perrine SP. Fetal globin stimulant therapies in the beta-hemoglobinopathies: principles and current potential. Pediatr Ann. 2008;37(5):339–346. doi: 10.3928/00904481-20080501-10. [DOI] [PubMed] [Google Scholar]
- 14.Perrine SP. Novel therapeutic agents for HbF induction: a new era for treatment of β thalassemia? Thalassemia Rep. 2011 doi: 10.4081/thal.2011.s2.e7. [DOI] [Google Scholar]
- 15.Neves F, Menezes Neto OA, Polis LB, Bassi SC, Brunetta DM, Silva-Pinto AC, et al. Hematological differences between patients with different subtypes of sickle cell disease on hydroxyurea treatment. Rev Bras Hematol Hemoter. 2012;34(6):426–429. doi: 10.5581/1516-8484.20120107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fathallah H, Atweh GF. DNA hypomethylation therapy for hemoglobin disorders: molecular mechanisms and clinical applications. Blood Rev. 2006;20(4):227–234. doi: 10.1016/j.blre.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Migliaccio AR, Rotili D, Nebbioso A, Atweh G, Mai A. Histone deacetylase inhibitors and hemoglobin F induction in β-thalassemia. Int J Biochem Cell Biol. 2008;40(11):2341–2347. doi: 10.1016/j.biocel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuccato C, Breda L, Salvatori F, Breveglieri G, Gardenghi S, Bianchi N, et al. A combined approach for β-thalassemia based on gene therapy-mediated adult hemoglobin (HbA) production and fetal hemoglobin (HbF) induction. Ann Hematol. 2012;91(8):1201–1213. doi: 10.1007/s00277-012-1430-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fibach E. Involvement of phosphatases in proliferation, maturation, and hemoglobinization of developing erythroid cells. J Signal Transduct. 2011 doi: 10.1155/2011/860985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rigano P, Pecoraro A, Calzolari R, Troia A, Acuto S, Renda D, et al. Desensitization to hydroxycarbamide following long-term treatment of thalassaemia intermedia as observed in vivo and in primary erythroid cultures from treated patients. Br J Haematol. 2010;151(5):509–515. doi: 10.1111/j.1365-2141.2010.08397.x. [DOI] [PubMed] [Google Scholar]
- 21.Thein SL. The emerging role of fetal hemoglobin induction in non-transfusion-dependent thalassemia. Blood Rev. 2012;26:S35–S39. doi: 10.1016/S0268-960X(12)70011-5. [DOI] [PubMed] [Google Scholar]
- 22.El-Beshlawy A, Hamdy M, El Ghamrawy M. Fetal globin induction in beta-thalassemia. Hemoglobin. 2009;33(S1):S197–S203. doi: 10.3109/03630260903351882. [DOI] [PubMed] [Google Scholar]
- 23.Bianchi N, Zuccato C, Lampronti I, Borgatti M, Gambari R. Fetal Hemoglobin inducers from the natural world: a novel approach for identification of drugs for the treatment of β-thalassemia and sickle-cell anemia. Evid Based Complement Alternat Med. 2009;6(2):141–151. doi: 10.1093/ecam/nem139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huisman TH. A short review of human γ-globin gene anomalies. Acta Haematol. 1987;78:80–84. doi: 10.1159/000205850. [DOI] [PubMed] [Google Scholar]
- 25.Singer K, Chernoff AI, Singer L. Studies on abnormal hemoglobins. II. Their identification by means of the method of fractional denaturation. Blood. 1951;6(5):429–435. [PubMed] [Google Scholar]
- 26.Atweh GF. Hydroxyurea in sickle cell disease: what will it take to change practice? Am J Hematol. 2010;85(6):401–402. doi: 10.1002/ajh.21733. [DOI] [PubMed] [Google Scholar]
- 27.Ware RE, Aygun B. Advances in the use of hydroxyurea. Hematology Am Soc Hematol Educ Program. 2009 doi: 10.1182/asheducation-2009.1.62. [DOI] [PubMed] [Google Scholar]
- 28.Banan M, Bayat H, Azarkeivan A, Mohammadparast S, Kamali K, Farashi S, et al. The XmnI and BCL11A single nucleotide polymorphisms may help predict hydroxyurea response in Iranian β-thalassemia patients. Hemoglobin. 2012;36(4):371–380. doi: 10.3109/03630269.2012.691147. [DOI] [PubMed] [Google Scholar]
- 29.Hashemi A, Abrishamkar M, Jenabzade AR, Eslami Z. Hydroxyurea can reduce or eliminate transfusion requirements in children with major and intermediate thalassemia. Iran J Blood Cancer. 2009;1(4):147–150. [Google Scholar]
- 30.Karimi M, Cohan N, Moosavizadeh K, Javad Falahi M, Haghpanah S. Adverse effects of hydroxyurea in β-thalassemia intermedia patients: 10 years' experience. Pediatr Hematol Oncol. 2010;27(3):205–211. doi: 10.3109/08880011003639952. [DOI] [PubMed] [Google Scholar]
- 31.Mokhtar GM, Tantawy AA, Adly AA, Ismail EA. Clinicopathological and radiological study of egyptian β-thalassemia intermedia and β-thalassemia major patients: relation to complications and response to therapy. Hemoglobin. 2011;35(4):382–405. doi: 10.3109/03630269.2011.598985. [DOI] [PubMed] [Google Scholar]
- 32.Bohara VV, Ray S, Chakrabarti P, Ray SS, Nath UK, Chaudhuri U. Optimizing the dose of hydroxyurea therapy for patients with β-thalassemia intermedia (Hb E-β-thalassemia): a single center study from eastern India. Hemoglobin. 2014;38(1):44–48. doi: 10.3109/03630269.2013.845844. [DOI] [PubMed] [Google Scholar]
- 33.Banerjee A, Chakrabarty SB, Karmakar SR, Chakrabarty A, Biswas SJ, Haque S, et al. Can homeopathy bring additional benefits to thalassemic patients on hydroxyurea therapy encouraging results of a preliminary study. Evid Based Complement Alternat Med. 2010;7(1):129–136. doi: 10.1093/ecam/nem161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meiler SE, Wade M, Kutlar F, Yerigenahally SD, Xue Y, Moutouh-de Parseval LA, et al. Pomalidomide augments fetal hemoglobin production without the myelosuppressive effects of hydroxyurea in transgenic sickle cell mice. Blood. 2011;118(4):1109–1112. doi: 10.1182/blood-2010-11-319137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehsani MA, Hedayati-Asl AA, Bagheri A, Zeinali S, Rashidi A. Hydroxyurea-induced hematological response in transfusion-independent beta-thalassemia intermedia: case series and review of literature. Pediatr Hematol Oncol. 2009;26(8):560–565. doi: 10.3109/08880010903271671. [DOI] [PubMed] [Google Scholar]
- 36.Kosaryan M, Vahidshahi K, Karami H, Ehteshami S. Effect of hydroxyurea on thalassemia major and thalassemia intermedia in Iranian patients. Pak J Med Sci. 2009;25(1):74–78. [Google Scholar]
- 37.Elalfy MS, Adly AA, Ismail EA, Elhenawy YI, Elghamry IR. Therapeutic superiority and safety of combined hydroxyurea with recombinant human erythropoietin over hydroxyurea in young β-thalassemia intermedia patients. Eur J Haematol. 2013;91(6):522–533. doi: 10.1111/ejh.12182. [DOI] [PubMed] [Google Scholar]
- 38.Karimi M, Mohammadi F, Behmanesh F, Samani SM, Borzouee M, Amoozgar H, et al. Effect of combination therapy of hydroxyurea with l-©\carnitine and magnesium chloride on hematologic parameters and cardiac function of patients with β-thalassemia intermedia. Eur J Haematol. 2010;84(1):52–58. doi: 10.1111/j.1600-0609.2009.01356.x. [DOI] [PubMed] [Google Scholar]
- 39.Rigano P, Pecoraro A, Calvaruso G, Steinberg MH, Iannello S, Maggio A. Cerebrovascular events in sickle cell-beta thalassemia treated with hydroxyurea: a single center prospective survey in adult Italians. Am J Hemato. 2013;88(11):E261–E264. doi: 10.1002/ajh.23531. [DOI] [PubMed] [Google Scholar]
- 40.Italia K, Jijina F, Merchant R, Swaminathan S, Nadkarni A, Gupta M, et al. Comparison of in-vitro and in-vivo response to fetal hemoglobin production and γ-mRNA expression by hydroxyurea in hemoglobinopathies. Indian J Hum Genet. 2013;19(2):251–258. doi: 10.4103/0971-6866.116128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dixit A, Chatterjee TC, Mishra P, Choudhry DR, Mahapatra M, Tyagi S, et al. Hydroxyurea in thalassemia intermedia—a promising therapy. Ann Hematol. 2005;84(7):441–446. doi: 10.1007/s00277-005-1026-4. [DOI] [PubMed] [Google Scholar]
- 42.Makropoulos DA, Achuthanandam R, Avery J, Wilson K, Brosnan K, Miller A, et al. CNTO 530 increases expression of HbA and HbF in murine models of β-thalassemia and sickle cell anemia. Curr Pharm Biotechnol. 2013;14(2):242–248. doi: 10.2174/138920113805219449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taher AT, Musallam KM, Karimi M, Cappellini MD. Contemporary approaches to treatment of beta-thalassemia intermedia. Blood Rev. 2012;26:S24–S27. doi: 10.1016/S0268-960X(12)70008-5. [DOI] [PubMed] [Google Scholar]
- 44.Zeng YT, Huang SZ, Ren ZR, Lu ZH, Zeng FY, Schechter AN, et al. Hydroxyurea therapy in β-thalassaemia intermedia: improvement in haematological parameters due to enhanced β-globin synthesis. Br J Haematol. 2008;90(3):557–563. doi: 10.1111/j.1365-2141.1995.tb05584.x. [DOI] [PubMed] [Google Scholar]
- 45.Li B, Ding L, Li W, Story MD, Pace BS. Characterization of the transcriptome profiles related to globin gene switching during in vitro erythroid maturation. BMC Genomics. 2012;13(1):153. doi: 10.1186/1471-2164-13-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng Y, Huang S. The studies of hemoglobinopathies and thalassemia in China—the experiences in Shanghai Institute of Medical Genetics. Clin Chim Acta. 2001;313(1–2):107–111. doi: 10.1016/s0009-8981(01)00660-x. [DOI] [PubMed] [Google Scholar]
- 47.Thein SL. Genetic modifiers of the beta-haemoglobinopathies. Br J Haematol. 2008;141(3):357–366. doi: 10.1111/j.1365-2141.2008.07084.x. [DOI] [PubMed] [Google Scholar]
- 48.Fucharoen S, Siritanaratkul N, Winichagoon P, Chowthaworn J, Siriboon W, Muangsup W, et al. Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E disease. Blood. 1996;87(3):887–892. [PubMed] [Google Scholar]
- 49.Haj Khelil A, Morinière M, Laradi S, Khelif A, Perrin P, Ben Chibani J, et al. Xmn I polymorphism associated with concomitant activation of Gγ and Aγ globin gene transcription on a β0-thalassemia chromosome. Blood Cells Mol Dis. 2011;46(2):133–138. doi: 10.1016/j.bcmd.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Pourfarzad F, von Lindern M, Azarkeivan A, Hou J, Kia SK, Esteghamat F, et al. Hydroxyurea responsiveness in β-thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity. Haematologica. 2013;98(5):696–704. doi: 10.3324/haematol.2012.074492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zamani F, Shakeri R, Eslami SM, Razavi SM, Basi A. Hydroxyurea therapy in 49 patients with major beta-thalassemia. Arch Iran Med. 2009;12(3):295–297. [PubMed] [Google Scholar]