

Abstract
We describe a phenotypic high throughput screening (HTS) calcium flux assay designed to identify pharmacoperones for the gonadotropin releasing hormone receptor (GnRHR). Pharmacoperones are target-specific, small molecules that diffuse into cells, rescue misfolded protein mutants, and restore them to function. Rescue is based on correcting the trafficking of mutants that would otherwise be retained in the endoplasmic reticulum and unable to function correctly. This approach identifies drugs with a significant degree of novelty, relying on cellular mechanisms that are not currently exploited. Development of such assays is important, since the extensive use of agonist/antagonist screens alone means that useful chemical structures may be present in existing libraries but have not been previously identified using existing methods. Our assay utilizes cell lines stably expressing a GnRHR mutant under the control of a tetracycline (OFF) transactivator. This allows us to quantitate the level of functional and properly trafficked G protein coupled receptors present in each test well. Furthermore, since we are able to turn receptor expression on and off, we can rapidly eliminate the majority of false positives from our screening results. Our data show that this approach is likely to be successful in identifying hits from large chemical libraries.
Introduction
G protein coupled receptors (GPCRs), which include the gonadotropin releasing hormone (GnRH) receptor (GnRHR), comprise the largest family of validated drug targets—35%–50% of approved drugs derive their benefits by selective targeting of GPCRs. High throughput screen (HTS) assays for drugs targeting GPCRs have been identified using screens for either agonists or antagonists. Our work and that of others shows that valuable drugs that affect the trafficking of GPCRs may have been overlooked because of this limitation.1,2
Mutations in GPCRs frequently result in misrouted proteins and are known to be responsible for more than 30 disorders, including cancers, heritable obesity, and endocrine disease. Included in this group is hypogonadotropic hypogonadism (HH) caused by mutations in the GnRHR,3–14 and other disorders of reproduction that result from inappropriately decreased plasma membrane expression of the WT receptor.15
The GnRHR resides in the gonadotrope cells of the pituitary and is responsible for producing responses to hypothalamic GnRH, such as the release of luteinizing hormone (LH) and follicle stimulating hormone (FSH). The human GnRHR (hGnRHR) has been a central focus of drug development, and understanding the mechanism of GnRH action has already led to useful drugs (agonists and antagonists).16
Normally, GPCRs are subjected to a stringent quality control system (QCS) in the endoplasmic reticulum (ER). This system consists of both protein chaperones that retain misfolded proteins and enzyme-like proteins that catalyze the folding process. The QCS (consisting of endogenous chaperones), which assesses structure but not function, ensures that only correctly folded proteins enter the pathway leading to the plasma membrane (PM). Because of this, point mutations may result in the production of misfolded and disease-causing proteins that are unable to reach their functional destinations in the cell because they are retained by the QCS even though they may retain (or regain) function.
The functional rescue of misfolded mutant receptors by small nonpeptide molecules (pharmacoperones), originally screened from libraries to serve as receptor antagonists, has now been demonstrated.17 A pharmacoperone is a small molecule that enters cells and serves as a “molecular scaffold” to promote correct folding of otherwise misfolded mutant proteins within the cell.8,18 Misfolded proteins are frequently retained by the cellular QCS of the ER, do not reach their normal site of function,19,20 and may result in disease.21 Pharmacoperones rescue misfolded receptor mutants and restore them to function, which is a potentially useful therapeutic approach when the target is a misfolded/misrouted protein.
We summarized the literature for the pharmacoperones of the GnRH with a view toward moving these compounds in vivo.17 Science writers commenting on this work22,23 have observed that rescue with pharmacoperones is a viable “alternative (to gene therapy),” since it serves as a means of “skirting gene therapy to correct genetic defects.” This view is supported by the consideration that correction of defective protein folding appears significantly less challenging than replacement of a defective gene (or gene product) by a perfect one. Protein rescue with pharmacoperones results from proper folding, passage through the QCS, restoration to the proper site, and return of function.
All pharmacoperone drugs identified to date for the GnRHR were identified from hits in HTS screens for receptor antagonists and repurposed. These must be removed after rescue to preclude competition with agonists. This results in a complex pharmacology for drug administration. This therapeutic problem will be addressed by identification of drug candidates in the proposed assays that are pharmacoperones but lack antagonistic activity.
Recently, we have developed a mouse phenotype expressing the E90K mutation of the GnRHR that causes HH in humans and mice. Conditions for pharmacoperone-rescue of this mutant in vivo have been established,24 and this animal establishes in vivo proof of principle for the efficacy of this class of drugs.
Materials and Methods
Materials
IN3 (((2S)-2-[5-[2-(2-azabicyclo[2.2.2]oct-2-yl)-1,1-dimethyl-2-oxoethyl] 2-(3,5-dimethylphenyl)-1H-indol-3-yl]-N-(2-pyridin-4-ylethyl) propan-1-amine)) and Q89 (7-chloro-2-oxo-4-{2-[(2S)-piperidin-2-yl]ethoxy}-N-pyrimidin-4-yl-3-(3,4,5-trimethylphenyl)-1,2-dihydroquinoline-6-carboxaminde), both positive control compounds, were a kind gift from Merck and Company and were discovered as peptidomimetic antagonists of the GnRH receptor. The pilot library (LOPAC) was obtained from Sigma-Aldrich (St. Louis, MO) and stored as 2.5 mM dimethylsulfoxide (DMSO) stock solutions at −20°C in sealed polypropylene plates. The stable cells were created in HeLa cells as previously described.25 The GnRH receptor agonist was synthesized and biologically characterized by us. Fetal bovine serum (FBS) was obtained from HyClone (Logan, UT).
HTS Optimized Primary GnRHR Pharmacoperone Assay
HeLa cells stably expressing GnRHR[E90K] under the control of a tetracycline-controlled transactivator were cultured in growth media (1× Dulbecco's modified Eagle's medium [DMEM] +10% FBS and 1 mg/L gentamicin) as described in Table 1. On the day of screening, cells were trypsinized and added to plates (20 μL/well, 8,500 cells/well). This was followed immediately by pin-tool addition (Kalypsys, San Diego, CA) of test compounds and controls (positive control was 100 nM IN3 or Q89 as specific; negative control was carrier only) in 100 nL DMSO. The drugged plates were incubated for 17 h at 37°C, 5% CO2 prior to addition of 20 μL of Fluo-2 dye. Following a 1 h incubation at 37°C, 5% CO2, and 30 min room temperature equilibration with dye, 5 μL of GnRH (500 nM final) was added in assay buffer (HBSS+20 mM HEPES+3% DMSO) followed by determination of calcium release by the FLIPR. We use a sensitive, nonradioactive, homogenous “mix-and-read” HTS assay protocol,26 which measures Ca2+ levels via a Fluo-2 a calcium sensitive cell permeable dye (Fig. 1). The optimized counterscreen was identical to the primary screen described except that cells were cultured in the presence of 1 μg/mL doxycycline for 36 h prior to plating and during all phases of the experiment.
Table 1.
Example of Gonadotropin Releasing Hormone Receptor High Throughput Screening Assay Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Plate cellsa | 20 μL | 8,500 HeLa GnRHR E90K cells |
| 2 | Pin compounds and control | 100 nL | IN3, DMSO |
| 3 | Incubate | Overnight | 37°C, 5% CO2 |
| 4 | Reporter dye | 20 μL | Fluo-2 dye |
| 5 | Incubate | 1 h | 37°C, 5% CO2 |
| 6 | Incubate | 30 min | Ambient temperature |
| 7 | Assay readout | 490ex/530em | Kinetic read for 140 sec |
Step Notes
1. Greiner 384-well part 781091 (Frickenhausen Germany).
2. Column 2 receives IN3 (high control); Column 23 receives DMSO (low control); columns 3–22 receive test compound; columns 1 and 24 receive cells.
3. Plates covered with stainless steel gasket lined lids containing pinholes for gas exchange.
4. Single tip dispense reagent all wells.
5. Plates covered with stainless steel gasket lined lids containing pinholes for gas exchange.
6. Plates lidded until moved to FLIPR Tetra (Molecular Devices).
7. Add 5 μL of GnRH (500 nM final) in assay buffer, or DMSO equivalent, and read using kinetic read modality on FLIPR tetra. Read settings include 470_495 LED excitation, 515_575 emission, gain of 140, exposure time of 0.40 and excitation intensity of 70. Export the data as Max signal obtained divided by the Min signal=Ratio (Max/Min).
For the primary assay, cells are not treated with doxycycline and hence are induced to express the GnRHR (per the Tet-off system). The counterscreen assay uses doxycycline-treated cells.
IN3, ((2S)-2-[5-[2-(2-azabicyclo[2.2.2]oct-2-yl)-1,1-dimethyl-2-oxoethyl] 2-(3,5-dimethylphenyl)-1H-indol-3-yl]-N-(2-pyridin-4-ylethyl) propan-1-amine); DMSO, dimethyl sulfoxide; GnRH, gonadotropin releasing hormone; GnRHR, GnRH receptor.
Fig. 1.

Gonadotropin releasing hormone (GnRH) receptor (GnRHR) pharmacoperone assay principle. The mutant GnRHR is retained in the lumen of untreated cells. Addition of pharmacoperone rescues GnRHR, and the receptors are trafficked to the plasma membrane. GnRHR on the plasma membrane is now responsive to agonism by GnRH, which is quantitated using Fluo-2 detection reagents (box). The level of functional GnRHR (mutant) is proportional to the magnitude of Gq modulated signaling, IP3 induction, and subsequent Ca2+ released and detected in the cells via Fluo2 using the FLIPR Tetra reader as illustrated via the kinetic traces shown in the lower inset diagram.
Data Processing
The raw fluorescence data were interpreted as a ratio of Max/Min values, which were normalized to positive and negative controls to give percent response scores. Dose–response curves were fit using a four-parameter variable slope sigmoidal curve in GraphPad Prism 5.02 (GraphPad Software, La Jolla, CA). Concentration response curves were generated using Graphpad software via a four-parameter equation, which yield sigmoidal concentration response curves fitted using nonconstrained upper and lower boundaries parameters and variable slope.
Results
Overview of the Assay
In this HTS, the level of functional GnRHR present in each test well is quantitated using a calcium sensitive dye FLIPR-based assay system (Fig. 1). This technique allows the screen to identify compounds that increase the trafficking of mutant GnRHR[E90K] in our model system. To triage assay artifact and compounds with intrinsic off-target activity, compounds are counterscreened with the same cell line as the primary assay, except in the presence of doxycycline, which shuts off the GnRHR[E90K] expression.
The GnRHR Mutant
We selected mutant GnRHR[E90K] as the basis of our screen; this mutant causes human HH. Modeling studies for the hGnRHR and experimental data support the view that the E90-K121 salt bridge is a fundamental and evolutionarily conserved determinant required for correct protein trafficking to the PM in all mammals examined.27–29 This bridge links transmembrane segment 2 (TMS2) to TMS3. Because this salt bridge is a requirement for correct routing, mutation E90K results in a routing defect in both mouse and human GnRHR.30,31 This leads to full but pharmacoperone rescuable ER-retention8 and the predicted phenotype in humans32 and mice.33
Pharmacoperone IN3 rescues mutants of the GnRHR
When cells expressing mutant GnRHR[E90K], which is recognized by the cellular quality control system as defective and retained in the ER, are incubated with IN3, the mutant is rescued.8,33–35 This pharmacoperone rescues many mutants of the hGnRHR, is GPCR-specific, and was used as a positive control in our assay development.34,36
The GnRHR pharmacoperone primary HTS and counterscreen
GnRHR[E90K] activity is coupled to Ca2+ flux. The primary assay for this project uses a HeLa cells constitutively expressing GnRHR[E90K] controlled by a tetracycline-regulated trans-activator.25,37 In the absence of doxycycline (a stable analog of tetracycline), the mutant is expressed; it is then misrouted and retained in the endoplasmic reticulum (ER).
Following pretreatment with pharmacoperone, the mutant is rescued and trafficked to the plasma membrane. The rescued mutant is then responsive to native GnRH and signals, via Gq coupling, resulting in the subsequent PLC-β activation leading to PIP2 hydrolysis into IP3. IP3 in turns affects the release of intracellular Ca2+, which is readily quantified via Fluo-2 dye using the FLIPR Tetra reader.
The counterscreen protocol is identical to the primary HTS assay protocol with the exception that the GnRHR[E90K] cells are incubated in the presence of 1 μg/mL of doxycycline (“Dox”) for 36 h prior to assay. During this time, the gene for the mutant is off, and no measurable mutant remains in the cell. Accordingly, the GnRHR[E90K] primary assay and counterscreen protocols confirm the expected pharmacology of positive and negative controls (Fig. 2). As a point of comparison, IN3 was originally designed as a GnRHR antagonist.38–40 The result with IN3 validates using the Dox+GnRHR[E90K] Fluo-2 assay as the counterscreen for identification and elimination of pharmacoperone hits that are also antagonists.
Fig. 2.

Responses of controls in the 384-well formatted GnRHR[E90K] and wild type pharmacoperone assay. (A) Mutant cells. In the absence of doxycycline (“Dox”), GnRHR[E90K] is synthesized and retained in the endoplasmic reticulum (ER). Following pretreatment with the pharmacoperone IN3, GnRHR[E90K] is rescued and trafficked to the plasma membrane, and a robust Ca2+ response to GNRH (500 nM) challenge is observed (●). Also included are the results of challenging the cells with GnRH, after preincubation with IN3 and doxycycline at 1 μg/mL (▲), as well as similar experiments without the GnRH challenge (■ and ▼). (B) Wild type cells. In the absence of Dox, wild type GnRHR is expressed and automatically trafficked to the plasma membrane. Upon GnRH challenge, the pharmacoperone IN3 decreases the amount of Ca2+ response in these cells, in a concentration-dependent manner, because IN3 acts as an antagonist in this format (●). Also shown are the control experiments in the wild type cells, similar to those done with the mutant cells. The data presented are means±standard deviation (sd) of quadruplicate wells (n=4).
The IN3 pharmacoperone yields an EC50 of 5.12±1.60 nM in the “-Dox” assay, and a signal-to-background ratio (S/B) of ∼2.5 (n=4). Both protocols are high throughput compatible in both 384- and 1,536-well formats, although the data presented here are obtained using a 384-well format.
An additional control compound of a different chemical class, Q89, was also tested and generated an EC50 of 1.9 nM (Fig. 3).
Fig. 3.

Q89 Rescues the mutant GnRHR[E90K]. Cells were grown, and Q89 was used at different doses to rescue GNRHR. The EC50 for Q89 was determined to be 1.9 nM (●) in the presence of GNRH (500 nM) and had no effect when GNRH was absent (■). The data presented are means±sd of quadruplicate wells (n=4).
LOPAC pilot screen
The Sigma LOPAC (Library of Pharmacologically Active 1280 Compounds) was screened to determine the performance of the optimized GnRHR[E90K] pharmacoperone assay in terms of robustness (Z′). Briefly, compounds were analyzed at a single concentration of nominally 6 μM (0.2% DMSO) using IN3 as a positive control. Each plate contained high and low signal control wells, which were used in Z′ factor calculations. An activity scatterplot of all compounds tested, as well as positive and negative controls, is shown in Figure 4A. As indicated from the positive and negative control scatterplots, the assay demonstrated an acceptable Z′ factor (0.57±0.06; n=12 plates) for the entire LOPAC screen, indicative of an assay that is amenable to HTS.
Fig. 4.

Scatter plot analysis of the high throughput LOPAC pilot screen. (A) All data from all assay plates (n=12, triplicate results), including controls, are displayed. The separation in activity (Z′=0.57±0.06, S/B=1.7±0.1) between wells dosed with IN3 (“high control”) and DMSO (“low control”) indicates a HTS assay that will allow for reproducible selection of hits. (B) Representative graph of IN3 pharmacoperone activity performed while running the LOPAC pilot screen. The EC50 (5.4 nM) indicates reproducible and expected assay sensitivity was achieved. The data presented are means±sd of quadruplicate wells (n=4).
Day-to-day reproducibility of the assay is also excellent as indicated by the control pharmacoperone IN3, which, met with the expected sensitivity during this LOPAC pilot HTS, is referenced in Figure 4B. Achieving passing Z values along with the expected pharmacology of the control allows us to proceed to hit analysis. In spite of having a low average activity for the tested compounds (results in triplicate) and using a nominal hit-cutoff (85) of 18.24% (average±3 SD of sample field), no hits were identified. This is not unexpected based on the limited number of compounds in the LOPAC collection and the complexity of the target. LOPAC is a small library containing no structures that are similar to any of the controls based on a Tanimoto score >90. Based on results from previous cell-based screens, we anticipate an actual HTS hit rate <1%.
Although the LOPAC screen yielded low activities, 10 compounds with the highest activities were reexamined and a subset with notable Dox−/Dox+ differentiation from their respective screens was found and is presented in Table 2.
Table 2.
Primary Assay and Counterscreen Results for Three of the Most Active Compounds from the LOPAC Screen
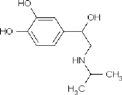
| Naftopidil dihydrochloride | (−)-Isoproterenol hydrochloride | (−)-Eseroline fumarate | |
|---|---|---|---|
| Structure | 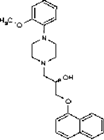 |
 |
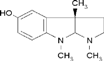 |
| Dox− response, max % (avg±SD) | 5.5±9.4 | 1.6±2.4 | 1.0±1.1 |
| Dox+ response, max % (avg±SD) | 0.4±1.6 | 4.0±4.0 | 3.3±5.4 |
| Vendor information | MicroSource 01506024 | Sigma-Aldrich I 2760 | Sigma-Aldrich E-100 |
| Selectivity | α1 Adrenoceptor antagonist; antihypertensive | β-Adrenoceptor agonist; increases cytosolic cAMP | Metabolite of physostigmine (eserine); potent analgesic; cholinesterase inhibitor |
avg, average; SD, standard deviation.
Confirmation of Mechanism of Action and Specificity
Compounds that show doxycycline-dependent signals will be profiled for specificity and potential mechanism of action using several plate-based high throughput assays. First, all hits will be tested for nonspecific cytotoxicity in unmodified human cell lines (BJ, HEPG2, and HEK293). Second, to determine if the observed pharmacoperone effect is specific to GnRHR or to a broader misrouted protein rescue phenomena, compounds showing activity in the GnRHR[E90K] Fluo-2 assay will be tested in our GnRHR[E90K] IP-One assay.25,37 This assay is identical to that for Fluo-2 assay, but measures the functional rescue of the ER-retained mutant E90K by quantitating levels of inositol phosphate one (IP1—a downstream marker for GnRHR activation) as an endpoint, rather than Ca2+. This assay has been optimized in 384-well format and published by our groups.25,37
In addition to these high throughput assays, a range of lower throughput approaches are available to characterize primary hits fully. The first of these is a well-characterized orthogonal endpoint, IP production, which has been in use in our group for more than 25 years41,42 and which shows good agreement with the high throughput assay system described above. This (IP) assay will identify artifacts arising from the Fluo-2 used in the primary HTS effort. Second, the ability of compounds to increase the amount of hGnRHR at the cell surface will be assessed using the well-established 125I-Buserelin (metabolically stable GnRH agonist) binding assay,43 as well as by monitoring cellular localization of GFP-tagged GnRHR[E90K] in transiently transfected COS-7 cells.36 GFP-tagged-GPCR approaches are well established and have been used in Conn's group and elsewhere for studying the localization of GnRHR.17,44 The combination of these approaches will allow us to confirm that primary hits are increasing levels of the downstream hGnRHR activation marker (IP production) by increasing the total amount of correctly folded receptor at the cell surface (increased 125I-Buserelin and decreased GFP-hGnRHR in the ER). Compounds able to do so will be classified as validated hits.
Discussion
For more than 20 years, there has been interest in the use of gene therapy to correct mutational disease. Issues related to the integration of therapeutic DNA into the genome, immune responses, technical problems with vectors (toxicity, immune, inflammatory responses, gene control, and targeting issues), chances of inducing tumors, (insertional mutagenesis), and other problems have made it challenging to reduce this approach to routine practice. Correcting the folding of misfolded protein mutants and restoring them to function (with pharmacoperone drugs) is a potential alternative to replacing them by gene therapy.
It is likely that valuable drugs reside in chemical libraries yet have been missed, since screening approaches that rely on identification of agonists and antagonists would have failed to identify pharmacoperone drugs. There are several advantages to using pharmacoperones, including the ability to restore misfolded proteins to function and not leave residual proteins behind that can result in activation of the unfolded protein response (UPR45), an event that causes other metabolic problems. One example of such problems is, if left unchecked, the UPR leads to cell death, an event believed to have evolved to remove unregulated cells from organisms.46 Another example includes the observations that in patients with retinitis pigmentosa, retinal cells undergo apoptosis due to retention of the causative mutant of rhodopsin,47 a GPCR. Further, in type 2 diabetes, β cells become damaged by elevated demand for insulin and UPR activation.48 Pharmacoperone drugs may provide a new way to accomplish this goal.
Pharmacoperone rescue potentially applies to a diverse array of human diseases that result from misfolding (these include cystic fibrosis,49–53 HH,8,54 nephrogenic diabetes insipidus,20,55,56 retinitis pigmentosa,57 hypercholesterolemia,58 cataracts,59 neurodegenerative diseases such as Alzheimer's, Huntington's, and Parkinson's,60–64 cancer,65 α1 trypsin deficiency and lysosomal storage disease,66,67 mucopolysaccharidosis type IIIC,68 and many others). One could envision drugs given in a prophylactic manner (e.g., in vitamins) that prevent the misfolding that leads to neurodegenerative disorders (Alzheimer's—misfolded amyloid69) Parkinson's (misfolded α-synuclein), and cataracts (misfolded lens crystalline). In this regard, diseases may be prevented before clinical signs present. In the case of certain proteins (e.g., the GnRHR, V2R, and rhodopsin), this approach has succeeded with a striking number of different mutants17 supporting the view that pharmacoperones will become powerful weapons in our therapeutic arsenal.17 Most pharmacoperones identified to date and all pharmacoperones of the GnRHR have been identified from screens that were developed to select antagonists. Accordingly, these drugs have both pharmacoperone and antagonist activity, which is therapeutically undesirable and presents a complex pharmacology. It has not been established whether binding at or near the binding site of the natural ligand is necessary for pharmacoperone activity, and there is existing information to suggest otherwise.35,70,71 This would, in fact, be an unexpected requirement, since one could imagine pharmacoperones that might stabilize the correctly routed form of the receptor and not show any antagonism (or agonism). Accordingly, identification of nonantagonistic pharmacoperones is a reasonable and therapeutically important goal. Further, this will provide insight into the mechanism of pharmacoperone action. These screens will identify therapeutic agents for human and animal disease and provide a much-needed framework and proof of principle for identification of pharmacoperone drugs for other GPRCs.
The physiology of GnRHR is well characterized in many animal models.72 A great deal of information is also available regarding the cellular mechanism of action and trafficking of the GnRHR.71 We have available substantive information on the mechanism of misfolding,27,29,35,36 mutant interactions with pharmacoperones,34–36 and the molecular basis of the dominant-negative effect.73,74
Two additional observations are important, since these extend the therapeutic potential of these drugs. First, pharmacoperone drugs need not be present at the time of protein synthesis, but can rescue ER-retained proteins that have already accumulated.36 This observation increases the therapeutic reach, since misfolded mutants need not be (first) degraded and then replaced by newly synthesized protein (i.e., the portion synthesized in the presence of pharmacoperone).
Second, while pharmacoperones are specific for individual proteins, those that rescue one mutant of an individual protein typically rescue most mutants of the same protein, likely by stabilizing a core region that makes the protein acceptable to the quality control system of the cell. This observation improves the therapeutic reach of these drugs,34 since each mutant of an individual protein will not require a separate drug.
We anticipate that data from identification of GnRHR pharmacoperones will provide a range of benefits. First, validated hits discovered will serve as the basis for developing potential therapeutic use in the treatment of HH.
Further, while GnRHRs is typically only expressed in the pituitary gonadotrope, it is also paradoxically expressed in virtually all melanomas, about 80% of human endometrial and ovarian cancers, and about 50% of breast cancers including triple-negative breast cancer, as well as bladder, colorectal, and pancreatic cancers, sarcomas, lymphomas, prostatic cancers, and renal cell carcinomas.75 For these cells, GnRH agonists are negative regulators of cancer growth. For example, activation of the GnRHR by exogenous agonists inhibits the proliferation of melanoma growth both in vitro and in vivo, indicating a direct antitumor activity of this class of compounds. Additionally, toxins conjugated to GnRH agonists are effectively targeted to melanoma cells where they show anti-angiogenic, anti-metastatic, and anti-oncogenic behavior.75–80 When GnRHR agonists or GnRH-toxin conjugates are used to treat melanoma, it is desirable to use the lowest dose consonant with therapeutic response, so as to limit side effects (i.e., androgen deprivation due to pituitary desensitization or nonspecific actions of the toxins). Pharmacoperones increase trafficking of the WT human GnRHR to the plasma membrane, a process that is otherwise about 50% efficient (i.e., about 50% is retained in the ER). Since selectively increasing the number of melanoma GnRHRs also increases the sensitivity of these cells to GnRH agonist, we expect that pharmacoperones will increase the sensitivity of these cells to GnRH agonist treatment, as well as to the toxin-GnRH conjugates. An additional use involves a subset of infertile women with responses to GnRH, suggesting a low plasma membrane expression of GnRHR.15,81 This is a candidate target for increased expression of WT GnRHR by pharmacoperones.
Control of anterograde (endoplasmic reticulum to plasma membrane) GnRHR trafficking is likely a key step in setting plasma membrane levels of this receptor, since it lacks binding sites for arrestin.82,83 Arrestin is associated with rapid retrograde trafficking (internalization).
In addition, compounds that more broadly affect protein folding and trafficking may be identified in this effort. The novel assay screening approach used in this project will provide a basis for other researchers interested in identifying small molecules that regulate protein trafficking.24
Abbreviations Used
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- FSH
follicle stimulating hormone
- GnRH
gonadotropin releasing hormone (pyroGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2)
- GnRHR
gonadotropin releasing hormone receptor
- GPCRs
G protein coupled receptors
- HH
hypogonadotropic hypogonadism
- HTS
high throughput screen
- IN3
((2S)-2-[5-[2-(2-azabicyclo[2.2.2]oct-2-yl)-1,1-dimethyl-2-oxoethyl] 2-(3,5-dimethylphenyl)-1H-indol-3-yl]-N-(2-pyridin-4-ylethyl) propan-1-amine)
- LH
luteinizing hormone
- Q89
(7-chloro-2-oxo-4-{2-[(2S)-piperidin-2-yl]ethoxy}designed-N-pyrimidin-4-yl-3-(3,4,5-trimethylphenyl)-1,2-dihydroquinoline-6-carboxaminde
- QCS
quality control system
- UPR
unfolded protein response
Disclosure Statement
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Acknowledgments
This work was supported by National Institutes of Health grants DK085040, OD012220, DK099090 (P.M.C.), and the National Institutes of Health's Roadmap Initiative grant U54MH084512 (E.S.,T.S., P.M.C., L.S.).
References
- 1.Conn PM, Ulloa-Aguirre A: Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol Metab 2010;21:190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conn PM, Janovick JA: Drug development and the cellular quality control system. Trends Pharmacol Sci 2009;30:228–233 [DOI] [PubMed] [Google Scholar]
- 3.Antelli A, Baldazzi L, Balsamo A, et al. : Two novel GnRHR gene mutations in two siblings with hypogonadotropic hypogonadism. Eur J Endocrinol 2006;155:201–205 [DOI] [PubMed] [Google Scholar]
- 4.Beranova M, Oliveira LM, Bedecarrats GY, et al. : Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2001;86:1580–1588 [DOI] [PubMed] [Google Scholar]
- 5.Bhangoo A, Jacobson-Dickman E: The genetics of idiopathic hypogonadotropic hypogonadism: unraveling the biology of human sexual development. Pediatr Endocrinol Rev 2009;6:395–404 [PubMed] [Google Scholar]
- 6.Chan YM, de Guillebon A, Lang-Muritano M, et al. : GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA 2009;106:11703–11708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E: Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 2003;100:10972–10976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janovick JA, Maya-Nunez G, Conn PM: Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab 2002;87:3255–3262 [DOI] [PubMed] [Google Scholar]
- 9.Leanos-Miranda A, Janovick JA, Conn PM: Receptor-misrouting: an unexpectedly prevalent and rescuable etiology in gonadotropin-releasing hormone receptor-mediated hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2002;87:4825–4828 [DOI] [PubMed] [Google Scholar]
- 10.Leanos-Miranda A, Janovick JA, Maya-Nunez G, Ulloa-Aguirre A, Conn PM: Misrouted proteins as a novel disease etiology and therapeutic target: rescue of hypogonadotropic hypogonadism-causing and manufactured mutants as a proof of principle. In: Molecular Endocrinology: Methods and Protocols. Kumar RA. (ed.), pp. 108–124 Humana Press, Totowa, NJ, 2003 [Google Scholar]
- 11.Leanos-Miranda A, Ulloa-Aguirre A, Janovick JA, Conn PM: In vitro coexpression and pharmacological rescue of mutant gonadotropin-releasing hormone receptors causing hypogonadotropic hypogonadism in humans expressing compound heterozygous alleles. J Clin Endocrinol Metab 2005;90:3001–3008 [DOI] [PubMed] [Google Scholar]
- 12.Maya-Nunez G, Janovick J, Aguilar-Rojas A, et al. : Biochemical mechanism of pathogenesis of human gonadotropin-releasing hormone receptor mutants Thr104Ile and Tyr108Cys associated with familial hypogonadotropic hypogonadism. Mol Cell Endocrinol 2011 2011;337:16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meysing AU, Kanasaki H, Bedecarrats GY, et al. : GNRHR mutations in a woman with idiopathic hypogonadotropic hypogonadism highlight the differential sensitivity of luteinizing hormone and follicle-stimulating hormone to gonadotropin-releasing hormone. J Clin Endocrinol Metab 2004;89:3189–3198 [DOI] [PubMed] [Google Scholar]
- 14.Seminara SB, Hayes FJ, Crowley WF, Jr: Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations. Endocr Rev 1998;19:521–539 [DOI] [PubMed] [Google Scholar]
- 15.Seminara SB, Beranova M, Oliveira LM, Martin KA, Crowley WF, Jr, Hall JE: Successful use of pulsatile gonadotropin-releasing hormone (GnRH) for ovulation induction and pregnancy in a patient with GnRH receptor mutations. J Clin Endocrinol Metab 2000;85:556–562 [DOI] [PubMed] [Google Scholar]
- 16.Conn PM, Crowley WF, Jr: Gonadotropin-releasing hormone and its analogues. N Engl J Med 1991;324:93–103 [DOI] [PubMed] [Google Scholar]
- 17.Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA: G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev 2007;59:225–250 [DOI] [PubMed] [Google Scholar]
- 18.Conn PM, Leanos-Miranda A, Janovick JA: Protein origami: therapeutic rescue of misfolded gene products. Mol Interv 2002;2:308–316 [DOI] [PubMed] [Google Scholar]
- 19.Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM: Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic 2004;5:821–837 [DOI] [PubMed] [Google Scholar]
- 20.Bernier V, Lagace M, Bichet DG, Bouvier M: Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab 2004;15:222–228 [DOI] [PubMed] [Google Scholar]
- 21.Castro-Fernandez C, Maya-Nunez G, Conn PM: Beyond the signal sequence: protein routing in health and disease. Endocr Rev 2005;26:479–503 [DOI] [PubMed] [Google Scholar]
- 22.Kresge N: Pharmacological chaperones show. Am Soc Biochem Molec Bio Today 2004;3:10–11 [Google Scholar]
- 23.Hunter PJ: Receptor redemption: skirting gene therapy to correct genetic defects. The Scientist 2004;18:30 [Google Scholar]
- 24.Janovick JA, Stewart MD, Jacob D, et al. : Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc Natl Acad Sci USA 2013;110:21030–21035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janovick JA, Park BS, Conn PM: Therapeutic rescue of misfolded mutants: validation of primary high throughput screens for identification of pharmacoperone drugs. PLoS One 2011;6:e22784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu K, Titus S, Southall N, Zhu P, Inglese J, Austin C, Zheng W: Comparison on functional assays for Gq-coupled GPCRs by measuring inositol monophospate-1 and intracellular calcium in 1536-well plate format. Curr Chem Genom 2008;1:70–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janovick JA, Conn PM: Salt bridge integrates GPCR activation with protein trafficking. Proc Natl Acad Sci USA 2010;107:4454–4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janovick JA, Patny A, Mosley R, et al. : Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Mol Endocrinol. 2009;23:157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janovick JA, Pogozheva ID, Mosberg HI, Conn PM: Salt bridges overlapping the gonadotropin-releasing hormone receptor agonist binding site reveal a coincidence detector for G protein-coupled receptor activation. J Pharmacol Exp Ther 2011;338:430–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janovick JA, Knollman PE, Brothers SP, Ayala-Yanez R, Aziz AS, Conn PM: Regulation of G protein-coupled receptor trafficking by inefficient plasma membrane expression: molecular basis of an evolved strategy. J Biol Chem 2006;281:8417–8425 [DOI] [PubMed] [Google Scholar]
- 31.Knollman PE, Janovick JA, Brothers SP, Conn PM: Parallel regulation of membrane trafficking and dominant-negative effects by misrouted gonadotropin-releasing hormone receptor mutants. J Biol Chem 2005;280:24506–24514 [DOI] [PubMed] [Google Scholar]
- 32.Soderlund D, Canto P, de la Chesnaye E, Ulloa-Aguirre A, Mendez JP: A novel homozygous mutation in the second transmembrane domain of the gonadotrophin releasing hormone receptor gene. Clin Endocrinol (Oxf) 2001;54:493–498 [DOI] [PubMed] [Google Scholar]
- 33.Janovick JA, Stewart MD, Jacob D, et al. : Restoration of plasma membrane expression and activity of a misfolded GnRHR mutant in mice by a small molecule pharmacoperone. Proc Natl Acad Sci USA 2013;110 [Google Scholar]
- 34.Janovick JA, Goulet M, Bush E, Greer J, Wettlaufer DG, Conn PM: Structure–activity relations of successful pharmacologic chaperones for rescue of naturally occurring and manufactured mutants of the gonadotropin-releasing hormone receptor. J Pharmacol Exp Ther 2003;305:608–614 [DOI] [PubMed] [Google Scholar]
- 35.Janovick JA, Patney A, Mosley R, et al. : Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Mol Endocrinol 2009;23:157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janovick JA, Brothers SP, Cornea A, et al. : Refolding of misfolded mutant GPCR: post-translational pharmacoperone action in vitro. Mol Cell Endocrinol 2007;272:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smithson DC, Janovick JA, Conn PM: Therapeutic rescue of misfolded/mistrafficked mutants: automation-friendly high throughput assays for identification of pharmacoperone drugs of GPCRs. Methods Enzymol 2013;521:3–16 [DOI] [PubMed] [Google Scholar]
- 38.Ashton WT, Sisco RM, Kieczykowski GR, et al. : Orally bioavailable, indole-based nonpeptide GnRH receptor antagonists with high potency and functional activity. Bioorg Med Chem Lett 2001;11:2597–2602 [DOI] [PubMed] [Google Scholar]
- 39.Ashton WT, Sisco RM, Yang YT, et al. : Substituted indole-5-carboxamides and -acetamides as potent nonpeptide GnRH receptor antagonists. Bioorg Med Chem Lett 2001;11:1723–1726 [DOI] [PubMed] [Google Scholar]
- 40.Ashton WT, Sisco RM, Yang YT, et al. : Potent nonpeptide GnRH receptor antagonists derived from substituted indole-5-carboxamides and -acetamides bearing a pyridine side-chain terminus. Bioorg Med Chem Lett 2001;11:1727–1731 [DOI] [PubMed] [Google Scholar]
- 41.Huckle WR, Conn PM: Use of lithium ion in measurement of stimulated pituitary inositol phospholipid turnover. Methods Enzymol 1987;141:149–155 [DOI] [PubMed] [Google Scholar]
- 42.Huckle WR, Conn PM: The relationship between gonadotropin-releasing hormone-stimulated luteinizing hormone release and inositol phosphate production: studies with calcium antagonists and protein kinase C activators. Endocrinology 1987;120:160–169 [DOI] [PubMed] [Google Scholar]
- 43.Marian J, Cooper RL, Conn PM: Regulation of the rat pituitary gonadotropin-releasing hormone receptor. Mol Pharmacol 1981;19:399–405 [PubMed] [Google Scholar]
- 44.Schulein R, Lorenz D, Oksche A, et al. : Polarized cell surface expression of the green fluorescent protein-tagged vasopressin V2 receptor in Madin Darby canine kidney cells. FEBS Lett 1998;441:170–176 [DOI] [PubMed] [Google Scholar]
- 45.Hetz C, Martinon F, Rodriguez D, Glimcher LH: The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev 2011;91:1219–1243 [DOI] [PubMed] [Google Scholar]
- 46.Walter P, Ron D: The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081–1086 [DOI] [PubMed] [Google Scholar]
- 47.Lin JH, Lavail MM: Misfolded proteins and retinal dystrophies. Adv Exp Med Biol 2010;664:115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fonseca SG, Gromada J, Urano F: Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol Metab 2011;22:266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kerem E: Pharmacological induction of CFTR function in patients with cystic fibrosis: mutation-specific therapy. Pediatr Pulmonol 2005;40:183–196 [DOI] [PubMed] [Google Scholar]
- 50.Dormer RL, Derand R, McNeilly CM, et al. : Correction of delF508-CFTR activity with benzo(c)quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci 2001;114:4073–4081 [DOI] [PubMed] [Google Scholar]
- 51.Galietta LJ, Springsteel MF, Eda M, et al. : Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J Biol Chem 2001;276:19723–19728 [DOI] [PubMed] [Google Scholar]
- 52.Zhang XM, Wang XT, Yue H, et al. : Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J Biol Chem 2003;278:51232–51242 [DOI] [PubMed] [Google Scholar]
- 53.Amaral MD: Therapy through chaperones: sense or antisense? Cystic fibrosis as a model disease. J Inherit Metab Dis 2006;29:477–487 [DOI] [PubMed] [Google Scholar]
- 54.Ulloa-Aguirre A, Janovick JA, Leanos-Miranda A, Conn PM: Misrouted cell surface GnRH receptors as a disease aetiology for congenital isolated hypogonadotrophic hypogonadism. Hum Reprod Update 2004;10:177–192 [DOI] [PubMed] [Google Scholar]
- 55.Bichet DG: Nephrogenic diabetes insipidus. Adv Chronic Kidney Dis 2006;13:96–104 [DOI] [PubMed] [Google Scholar]
- 56.Bernier V, Morello JP, Zarruk A, et al. : Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol 2006;17:232–243 [DOI] [PubMed] [Google Scholar]
- 57.Noorwez SM, Malhotra R, McDowell JH, Smith KA, Krebs MP, Kaushal S: Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem 2004;279:16278–16284 [DOI] [PubMed] [Google Scholar]
- 58.Wang QY, Manicassamy B, Yu X, Dolmer K, Gettins PG, Rong L: Characterization of the LDL-A module mutants of Tva, the subgroup A Rous sarcoma virus receptor, and the implications in protein folding. Protein Sci 2002;11:2596–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benedek GB, Pande J, Thurston GM, Clark JI: Theoretical and experimental basis for the inhibition of cataract. Prog Retin Eye Res 1999;18:391–402 [DOI] [PubMed] [Google Scholar]
- 60.Heiser V, Scherzinger E, Boeddrich A, et al. : Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington's disease therapy. Proc Natl Acad Sci USA 2000;97:6739–6744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Forloni G, Terreni L, Bertani I, et al. : Protein misfolding in Alzheimer's and Parkinson's disease: genetics and molecular mechanisms. Neurobiol Aging 2002;23:957–976 [DOI] [PubMed] [Google Scholar]
- 62.Permanne B, Adessi C, Saborio GP, et al. : Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer's disease by treatment with a beta-sheet breaker peptide. FASEB J 2002;16:860–862 [DOI] [PubMed] [Google Scholar]
- 63.Soto C, Kascsak RJ, Saborio GP, et al. : Reversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet 2000;355:192–197 [DOI] [PubMed] [Google Scholar]
- 64.Muchowski PJ, Wacker JL: Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 2005;6:11–22 [DOI] [PubMed] [Google Scholar]
- 65.Lubin DJ, Butler JS, Loh SN: Folding of tetrameric p53: oligomerization and tumorigenic mutations induce misfolding and loss of function. J Mol Biol 2010;395:705–716 [DOI] [PubMed] [Google Scholar]
- 66.Fan JQ: A contradictory treatment for lysosomal storage disorders: inhibitors enhance mutant enzyme activity. Trends Pharmacol Sci 2003;24:355–360 [DOI] [PubMed] [Google Scholar]
- 67.Bottomley SP: The structural diversity in alpha1-antitrypsin misfolding. EMBO Rep 2011;12:983–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feldhammer M, Durand S, Pshezhetsky AV: Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS One 2009;4:e7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun Y, Breydo L, Makarava N, Yang Q, Bocharova OV, Baskakov IV: Site-specific conformational studies of prion protein (PrP) amyloid fibrils revealed two cooperative folding domains within amyloid structure. J Biol Chem 2007;282:9090–9097 [DOI] [PubMed] [Google Scholar]
- 70.Janovick JA, Maya-Nunez G, Ulloa-Aguirre A, et al. : Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol Cell Endocrinol 2009;298:84–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Conn PM, Janovick JA: Trafficking and quality control of the gonadotropin releasing hormone receptor in health and disease. Mol Cell Endocrinol 2009;299:137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knobil E, Neill J, eds: The Physiology of Reproduction. Vols 1 & 2 Raven Press, New York, 1994 [Google Scholar]
- 73.Brothers SP, Cornea A, Janovick JA, Conn PM: Human loss-of-function gonadotropin-releasing hormone receptor mutants retain wild-type receptors in the endoplasmic reticulum: molecular basis of the dominant-negative effect. Mol Endocrinol 2004;18:1787–1797 [DOI] [PubMed] [Google Scholar]
- 74.Leanos-Miranda A, Ulloa-Aguirre A, Ji TH, Janovick JA, Conn PM: Dominant-negative action of disease-causing gonadotropin-releasing hormone receptor (GnRHR) mutants: a trait that potentially coevolved with decreased plasma membrane expression of GnRHR in humans. J Clin Endocrinol Metab 2003;88:3360–3367 [DOI] [PubMed] [Google Scholar]
- 75.Engel J, Emons G, Pinski J, Schally AV: AEZS-108: a targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin Investig Drugs 2012;21:891–899 [DOI] [PubMed] [Google Scholar]
- 76.Kovacs M, Schally AV, Nagy A, Koppan M, Groot K: Recovery of pituitary function after treatment with a targeted cytotoxic analog of luteinizing hormone-releasing hormone. Proc Natl Acad Sci USA 1997;94:1420–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Comaru-Schally AM, Brannan W, Schally AV, Colcolough M, Monga M: Efficacy and safety of luteinizing hormone-releasing hormone antagonist cetrorelix in the treatment of symptomatic benign prostatic hyperplasia. J Clin Endocrinol Metab 1998;83:3826–3831 [DOI] [PubMed] [Google Scholar]
- 78.Limonta P, Moretti RM, Marelli MM, Dondi D, Parenti M, Motta M: The luteinizing hormone-releasing hormone receptor in human prostate cancer cells: messenger ribonucleic acid expression, molecular size, and signal transduction pathway. Endocrinology 1999;140:5250–5256 [DOI] [PubMed] [Google Scholar]
- 79.Keller G, Schally AV, Gaiser T, et al. : Human malignant melanomas express receptors for luteinizing hormone releasing hormone allowing targeted therapy with cytotoxic luteinizing hormone releasing hormone analogue. Cancer Res 2005;65:5857–5863 [DOI] [PubMed] [Google Scholar]
- 80.Limonta P, Montagnani Marelli M, Mai S, Motta M, Martini L, Moretti RM: GnRH receptors in cancer: from cell biology to novel targeted therapeutic strategies. Endocr Rev 2012;33:784–811 [DOI] [PubMed] [Google Scholar]
- 81.Waldstreicher J, Seminara SB, Jameson JL, et al. : The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab 1996;81:4388–4395 [DOI] [PubMed] [Google Scholar]
- 82.Heding A, Vrecl M, Hanyaloglu AC, Sellar R, Taylor PL, Eidne KA: The rat gonadotropin-releasing hormone receptor internalizes via a beta-arrestin-independent, but dynamin-dependent, pathway: addition of a carboxyl-terminal tail confers beta-arrestin dependency. Endocrinology 2000;141:299–306 [DOI] [PubMed] [Google Scholar]
- 83.Hislop JN, Caunt CJ, Sedgley KR, et al. : Internalization of gonadotropin-releasing hormone receptors (GnRHRs): does arrestin binding to the C-terminal tail target GnRHRs for dynamin-dependent internalization? J Mol Endocrinol 2005;35:177–189 [DOI] [PubMed] [Google Scholar]