

Abstract
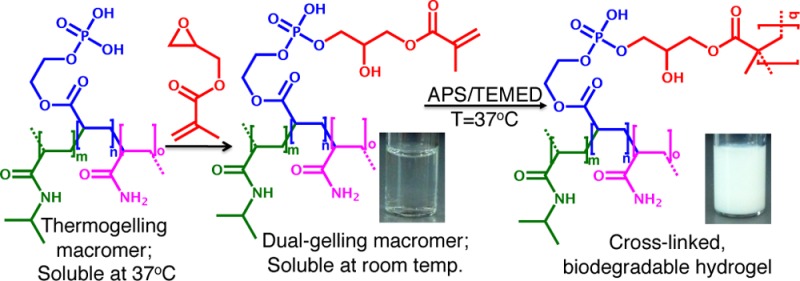
Novel, injectable, biodegradable macromer solutions that form hydrogels when elevated to physiologic temperature via a dual chemical and thermo-gelation were fabricated and characterized. A thermogelling, poly(N-isopropylacrylamide)-based macromer with pendant phosphate groups was synthesized and subsequently functionalized with chemically cross-linkable methacrylate groups via degradable phosphate ester bonds, yielding a dual-gelling macromer. These dual-gelling macromers were tuned to have transition temperatures between room temperature and physiologic temperature, allowing them to undergo instantaneous thermogelation as well as chemical gelation when elevated to physiologic temperature. Additionally, the chemical cross-linking of the hydrogels was shown to mitigate hydrogel syneresis, which commonly occurs when thermogelling materials are raised above their transition temperature. Finally, degradation of the phosphate ester bonds of the cross-linked hydrogels yielded macromers that were soluble at physiologic temperature. Further characterization of the hydrogels demonstrated minimal cytotoxicity of hydrogel leachables as well as in vitro calcification, making these novel, injectable macromers promising materials for use in bone tissue engineering.
Introduction
Hydrogels are promising materials for tissue engineering due to their highly hydrated environment, which facilitates exchange of nutrients and waste materials. Consequently, hydrogels can be used to deliver and support cells that can aid in tissue regeneration.1 Moreover, polymers that physically cross-link (thermogel) in response to changes in temperature to form hydrogels can be very useful for generating scaffolds in situ. These materials transition from a solution to a hydrogel at their lower critical solution temperature (LCST). When this temperature is between room temperature and physiologic temperature, these solutions have the potential to encapsulate cells and or growth factors as they are formed in situ upon reaching physiologic temperature following injection. Materials that are formed in situ also have the added benefit of being able to fill defects of all shapes and sizes.2,3
One commonly investigated group of synthetic thermogelling polymers is poly(N-isopropylacrylamide) (p(NiPAAm))-based polymers. P(NiPAAm) solutions undergo a near instantaneous phase transition at around 32 °C to form hydrogels. This transition temperature can be shifted by the incorporation of other monomers to form copolymers.4 However, it should be noted that p(NiPAAm)-based gels undergo postgelation syneresis, slowly deswelling and collapsing at temperatures above their LCST.5 This collapse can result in a significant expulsion of water, which removes many of the benefits of the hydrogel system. In an effort to mitigate this collapse, thermogelling macromers (TGMs) have been chemically cross-linked after thermogelation before the collapse can occur.5,6 This allows the benefit of the instantaneous gelation that occurs during thermogelation, as well as the hydrogel stability imparted by chemical cross-linking. Moreover, the amount of potentially cytotoxic chemically cross-linkable groups is decreased compared to gels that form completely via monomer polymerization in situ. Furthermore, dual-gelling macromers have been shown to support stem cell encapsulation, making them promising candidates for tissue engineering.7 However, one of the major pitfalls of many p(NiPAAm)-based hydrogels is that the copolymer backbones are nondegradable and, consequently, are not readily cleared from the body. In an effort to address this problem, side groups that become more hydrophilic upon hydrolytic,8,9 or catalytic10 degradation have been used to increase LCSTs of degraded TGMs above physiologic temperature allowing for the macromers to go back into solution.
We hypothesized that chemical cross-linking following thermogelation could be combined with hydrolysis-dependent LCST elevation, yielding in situ-forming, degradable hydrogels that have potential for use as cell-delivery vehicles. Specifically, phosphate esters were chosen for TGM LCST modulation via removal of hydrophobic groups. In addition to hydrolytic degradation, many phosphate esters can readily undergo catalytic degradation by alkaline phosphatase,11 which is commonly expressed in bone cells. This could accelerate hydrogel degradation as ALP-producing bone cells become more prevalent within the gels, secondary to either encapsulated cell differentiation or adjacent bone cell infiltration. Incorporation of phosphate groups into hydrogels has previously been shown to increase mineralization and improve function of encapsulated osteoblasts in bone tissue engineering applications.12,13
The objective of this study was to synthesize and characterize novel, injectable, thermoresponsive, phosphorus-containing, chemically cross-linkable macromers that form biodegradable hydrogels in situ. To accomplish these characteristics, NiPAAm was copolymerized with monoacryloxyethyl phosphate (MAEP) and acrylamide (AAm) to form TGMs with LCSTs above physiologic temperature. A factorial study was used to elucidate the effect of incorporation of the different monomers on the LCST. We hypothesized that the phosphate group of MAEP could be used to facilitate postpolymerization attachment of hydrophobic, chemically cross-linkable groups via degradable phosphate ester bonds, resulting in a decrease in LCST below physiologic temperature. Moreover, we hypothesized that the degradation of the phosphate ester bonds would yield a TGM with an LCST above physiologic temperature, resulting in soluble hydrogel degradation products.
Based on the results of the factorial study, two formulations with differing molar feeds of MAEP were selected for hydrogel characterization based on potential to be used for in vivo applications. Formulations were chosen so that they would have a transition temperature slightly below physiologic temperature following esterification, to allow for rapid thermogelation, as well as a transition temperature above physiologic temperature after degradation, to yield soluble degradation products. We hypothesized that chemical cross-linking of the hydrogel would mitigate syneresis. Additionally, the degradation, cytotoxicity, and in vitro mineralization of these hydrogel formulations were evaluated.
Methods
Materials
NiPAAm, AAm, azobis(isobutyronitrile) (AIBN), glycidyl methacrylate (GMA), glycerol, Tris-hydrochloride, magnesium chloride, zinc chloride, dimethyl sulfoxide (DMSO), D2O with 0.75 wt % 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid, sodium salt (TMP), sodium phosphate dibasic, butylated hydroxytoluene (BHT), ammonium persulfate (APS), tetramethylethylenediamine (TEMED), acetic acid, β-glycerol 2-phosphate, dexamethasone, ampicillin, amphotericin, and gentamicin were purchased from Sigma-Aldrich (St. Louis, MO) and used as received unless otherwise noted. MAEP was purchased from Polysciences Inc. (Warrington, PA). The solvents diethyl ether, acetone (analytical grade), and ethanol (200 proof) were obtained from VWR (Radnor, PA). Poly(ethylene glycol) (PEG) and poly(ethylene oxide) (PEO) standards were purchased from American Polymer (Mentor, OH). ALP from bovine intestinal mucosa (Sigma A2356) was diluted to 200 U/L in a buffered glycerol solution (50% glycerol, 50% 10 mM Tris-hydrochloride, 5 mM MgCl2, 0.2 mM ZnCl2, pH = 8.0) in accordance with the manufacturer’s protocol and was stored at 4 °C until used. Phosphate-buffered saline (PBS) solution was made from powder (pH 7.4, Gibco Life, Grand Island, NY), and ultrapure water was obtained from a Millipore Super-Q water system (Millipore, Billerica, MA). Complete osteogenic medium was made from minimal essential medium α (αMEM; Gibco Life, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Cambrex BioScience, Walkersville, MD), 10–8 M dexamethasone, 10 mM β-glycerol 2-phosphate, 50 mg/L ascorbic acid, 100 mg/L ampicillin, 250 mg/L amphotericin, and 50 mg/L gentamicin). Live/dead viability/cytotoxicity kit was purchased from Molecular Probes, Eugene, OR. The calcium assay was purchased from Genzyme Diagnostics, Cambridge, MA.
Macromer Synthesis
Statistical copolymers were synthesized from NiPAAm, AAm, and MAEP via free radical polymerization initiated by AIBN at 65 °C (Scheme 1). TGMs of the desired compositions were obtained by dissolving the monomers at the desired molar ratios (monomer feed) in DMSO, N2 purging of solution for 15 min, followed by heating the solution to 65 °C under a nitrogen atmosphere. Once the solution reached 65 °C, AIBN at a final concentration of 0.01 M was used to initiate the polymerization. In a typical experiment, 0.02 total moles of the corresponding monomers were dissolved in DMSO at 0.7 M. After AIBN injection, the reaction was stirred continuously at 65 °C for 20 h under a nitrogen atmosphere. The product was then concentrated via DMSO removal by rotoevaporation at 55 °C and 1 mbar, and redissolved in an 85/15 (v/v) mixture of acetone/DMSO at 9 mL/g starting material. This solution was added dropwise to cold diethyl ether to precipitate the copolymer while leaving unreacted monomers, initiators, and low molecular weight oligomers, in solution. Following vacuum filtration, the filtrate (a fine, white powder) was vacuumed dried at ambient temperature.
Scheme 1. Thermogelling Macromer (TGM) Formation.

TGMs were synthesized from the monomers N-isopropylacrylamide (NiPAAm), monoacryloxyethyl phosphate (MAEP), and acrylamide (AAm) by azobis(isobutyronitrile) (AIBN)-initiated free radical polymerization in dimethyl sulfoxide (DMSO).
Factorial Design
The thermogelling macromers were synthesized with high and low monomer levels to yield a 2 × 2 full factorial design (Table 1). The main effects and interaction of two variables (MAEP and AAm level) on LCST were examined. The high and low levels of MAEP listed in Table 2 were chosen to be similar to what has previously shown to improve in vitro mineralization of hydrogels made up of acrylic copolymers.14 The high and low levels of AAm listed in Table 2 were chosen to be in a range that would yield LCSTs above physiologic temperature based on preliminary experiments.
Table 1. Combinations of the Experimental Levels Used in the Factorial Designa.
| group | AAm | MAEP |
|---|---|---|
| 1 | – | – |
| 2 | + | – |
| 3 | – | + |
| 4 | + | + |
High (+) and low (−) levels of the monomers acrylamide (AAm) and monoacryloxyethyl phosphate (MAEP) are listed in Table 2.
Table 2. High (+) and Low (−) Levels for Monomers Acrylamide (AAm) and Monoacryloxyethyl Phosphate (MAEP) Used in the Factorial Design.
| AAm | MAEP | |
|---|---|---|
| high level | 18% | 12% |
| low level | 12% | 8% |
Macromer Methacrylation
Methacrylated TGMs (MA-TGMs) were synthesized via the esterification of phosphate groups of the TGMs with GMA, as shown in Scheme 2. In a typical reaction, 10 molar equivalents of GMA for every available P–OH group on the copolymer were added, with continuous stirring, to a mixture of vacuum-dried TGM and 5000 ppm BHT, a radical scavenger, at ambient temperature. This was immediately followed by the addition of ethanol at 2 mL/mg TGM. The reaction flask was stirred at ambient temperature for 10 min to allow the TGM to dissolve, then shielded from light, heated to 65 °C and stirred continuously for 40 h. The solution was allowed to cool to ambient temperature, diluted with an additional 3.5 mL ethanol/mg TGM, precipitated in diethyl ether, and vacuum filtered. The MA-TGM filtrate (a fine white powder) was dried under vacuum at ambient temperature.
Scheme 2. Methacrylated Thermogelling Macromer (MA-TGM) Formation.

MA-TGMs were formed via esterification of thermogelling macromers (TGMs) with glycidyl methacrylate (GMA) in ethanol. Butylated hydroxytoluene (BHT) was used as a free radical scavenger.
Proton Nuclear Magnetic Resonance (1H NMR) Spectroscopy
1H NMR spectroscopy was used to analyze the chemical composition of the copolymers. In a typical experiment, 20 mg of the TGM or MA-TGM were dissolved in 1 mL of D2O that contained 0.75 wt % TMP as an internal shift standard. Na2HPO4 ([10 mM]) was added to buffer the acidic TGM solutions and improve solubility in D2O at ambient temperature. Spectra were recorded at ambient temperature using a 400 MHz spectrometer (Bruker, Switzerland) and processed with TOPSPIN 3.0 (Bruker). To determine the composition of the TGMs, the spectra were integrated from 0.9 to 1.28 ppm (integral I1), 1.28–2.6 ppm (integral I2), and 3.61–4.60 ppm (integral I3), which were attributed to the protons for each group, as described in Figure 1A. These values were used to calculate the copolymer composition. TGM conversion to MA-TGM was determined by the ratio of the peaks from the hydrogens on the vinyl groups (5.63–5.85 ppm (integral I4) and 6.08–6.29 ppm (integral I5)) to the methyl groups (integral I1) of the NiPAAm monomer that was incorporated into the TGM (Figure 1B). We assumed that the molar composition of the copolymer backbone did not change upon methacrylation with GMA.
Figure 1.

Representative 1H NMR spectra of (A) a thermogelling macromer (TGM) and (B) a methacrylated thermogelling macromer (MA-TGM). Spectra were integrated from 0.9 to 1.28 ppm (integral I1), 1.28–2.6 ppm (integral I2), 3.61–4.60 ppm (integral I3), 5.63–5.85 ppm (integral I4), and 6.08–6.29 ppm (integral I5) to determine copolymer composition, with 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid, sodium salt (TMP) as an internal shift standard.
Differential Scanning Calorimetry (DSC)
A TA Instruments (New Castle, DE) differential scanning calorimeter was used to determine the LCST of the TGMs and MA-TGMs. In a typical experiment, 15 mg of TGM or MA-TGM were dissolved in 150 μL of PBS and 15 μL of the solution were placed in a DSC hermetic sample pan, which was then capped and crimped. Thermograms were recorded on a TA Instruments DSC 2920 against an empty pan as a reference. During a run, the oven was equilibrated at −5 °C for 10 min and then heated at a rate of 5 °C/min up to 80 °C. The LCST of the solution was determined as the maximum of the endothermic peak in the thermogram (endothermic up) using the Universal Analysis 2000 software provided by the DSC system. The LCSTs were expressed as means ± standard deviation (n = 3). The LCST values were analyzed by analysis of variance (ANOVA) with posthoc analysis by Tukey’s honestly significant different (HSD) test. Tests were conducted with a 95% confidence interval (α = 0.05). Main and interaction effects were analyzed using a linear regression analysis methodology via the SAS JMP Pro 10 software according to previously established methods.15
Size Exclusion Chromatography (SEC)
A gel permeation chromatography system made up of an HPLC pump (Waters, model 510, Milford, MA), an autosampler/injector (Waters, model 717), and a differential refractometer (Waters, model 410) with an Ultrahydrogel Linear SEC column (Waters, Part No. WAT011545) was used to determine the molecular weights and distributions of the synthesized copolymers. Solutions of copolymer were prepared at a concentration of 9 mg/mL in the mobile phase solvent and run in triplicate. Sample elution times in a 0.1 M NaNO3 mobile phase were used to determine number-average molecular weight (Mn) and polydispersity index (PDI) relative to PEG and PEO standards.
TGM Degradation
In order to characterize the LCST of degraded TGMs, 0.4 ALP units were added to TGM DSC samples prepared as described in the previous section and the samples were stored on a shaker table for 12 days at 37 °C to allow for hydrolysis of the phosphate ester bonds. In preliminary experiments taken out to 24 days, no further changes in LCST were seen after day 12 (data not shown). Following hydrolysis, samples were evaluated with DSC as described above.
Hydrogel Formation
MA-TGM solutions were prepared in PBS to give a final concentration of 15% (w/v) after the initiator volume was added. Stock solutions of the initiator system in PBS (pH 7.4) were added to the chilled MA-TGM solution to result in final APS and TEMED concentrations of 20 mM. The mixture was lightly agitated and 75 μL were pipetted into Teflon molds (7 mm diameter, 2 mm height). The molds were incubated at 37 °C for 2 h to allow the TGMs to thermally and chemically cross-link. After fabrication, the hydrogels were placed in PBS and stored at 37 °C. For experiments involving cell culture medium, the dried MA-TGMs were sterilized with UV radiation for 1 h prior to dissolution in sterile-filtered PBS and placed in medium following fabrication. No change in composition or release of small molecules due to bond cleavage was visualized in 1H NMR analysis of irradiated samples (data not shown).
Swelling Ratio Measurements
The swelling ratio was evaluated according to established protocols.7 At the desired time points, the gels were removed from the PBS and weighed (swollen weight). The hydrogels were then dried in a lyophilizer overnight and weighed (dry weight). The swelling ratio was calculated as (swollen weight-dry weight)/(dry weight). Swelling ratio was expressed as means and standard deviations (n = 5). The values were analyzed by ANOVA with posthoc analysis by Tukey’s HSD test. Tests were conducted with a 95% confidence interval (α = 0.05).
Hydrogel Degradation
After fabrication, the hydrogels were weighed and placed in 0.5 mL PBS (pH = 7.4) with or without 200 U/mL ALP and stored on a shaker table at 37 °C. The buffer was changed every 2–3 days to maintain pH. At the desired time points, hydrogels were removed from the buffer, weighed, and returned to buffer solution. Normalized weight was tracked over time. Normalized weight was expressed as means and standard deviations (n = 3), and values were analyzed by ANOVA with posthoc analysis by Tukey’s HSD test at each time point. Tests were conducted with a 95% confidence interval (α = 0.05).
Fourier Transform Infrared (FTIR) Spectroscopy
Following day 28 of the degradation study, hydrogels were rinsed with PBS, and dried in a lyophilizer. Dried samples from the degradation study and the swelling ratio study (24 h in PBS before being lyophilized) were analyzed with a Nicolet FTIR microscope. Spectra from two samples from each group were averaged and the spectra were normalized to have maximum transmittance of 100%.
Hydrogel Mineralization
Following fabrication, hydrogels were placed in complete osteogenic cell culture medium. Medium was changed every 2–3 days. At the desired time points, the hydrogels were removed from medium, rinsed with PBS, and weighed. The hydrogels were then placed in 500 μL of ultrapure water, and were manually homogenized. The suspensions then underwent three freeze–thaw cycles by alternately immersing in water at ambient temperature and liquid nitrogen, followed by probe ultrasonication for 5 s. Aliquots were then taken and mixed in equal parts with 1 N acetic acid (final concentration 0.5 N acetic acid) and incubated on a shaker table overnight at ambient temperature to dissolve the deposited calcium salts. The assay was performed according to the manufacturer’s instructions. All samples were run in triplicate and normalized to hydrogels that were not exposed to complete osteogenic cell culture medium. The data are expressed as means and standard deviations (n = 4) and values were analyzed by ANOVA with posthoc analysis by Tukey’s HSD test. Tests were conducted with a 95% confidence interval (α = 0.05).
Cell Culture
A rat fibroblast cell line (American Type Culture Collection no. CRL-1764) was cultured in cell culture medium (DMEM supplemented with 10% fetal bovine serum (FBS), 10 mM β-glycerol 2-phosphate, 50 mg/L ascorbic acid, 100 mg/L ampicillin, 250 mg/L amphotericin, and 50 mg/L gentamicin). The fibroblasts were cultured in a humidified incubator at 37 °C and 5% CO2. Cells of passage number 4 were used in this study.
Cytotoxicity of Hydrogel Leachables
The cytotoxicity of the dual-gelled hydrogels was evaluated by a leachables extraction test, in accordance with established protocols.16 Following fabrication, hydrogels were placed in cell culture medium at surface area:fluid volume ratio of 3 cm2/mL and incubated for 24 h at 37 °C. Following incubation, the hydrogels were removed from the supernatant, and 1×, 10×, and 100× dilutions were made with cell culture medium.
Cells were seeded on a 96-well plate at 80000 cells/mL and incubated in cell culture medium until 90% confluence was reached. The cell culture medium was then replaced with 100 μL of the hydrogel-conditioned media (n = 6/group). Live and dead controls were incubated in cell-culture medium with no exposure to the hydrogels. At the desired time points, media was removed, the dead controls were exposed to 70% ethanol for 10 min, and the cells were rinsed with PBS and then incubated for 30 min at ambient temperature in PBS containing calcein AM (2 μM) and ethidium homodimer-1 (4 μM) in accordance with the Live/Dead viability/cytotoxicity kit instructions. Cell viability was then quantified using a fluorescence plate reader (Biotek Instrument FLx800, Winooski, VT) equipped with filter sets of 485/528 nm (excitation/emission) for calcein AM (live cells) and 528/620 nm (excitation/emission) for ethedium homodimer-1 (dead cells). The fluorescence of the cell populations was recorded and the fractions of live and dead cells were calculated in accordance with the manufacturer’s instructions. The data are expressed as means and standard deviations (n = 6) and values were analyzed by ANOVA with posthoc analysis by Tukey’s HSD test. Tests were conducted with a 95% confidence interval (α = 0.05).
Results
TGM Synthesis and Characterization
The primary design criteria for the composition of the TGMs was the presence of thermoresponsive domains (NiPAAm), incorporation of phosphate groups (MAEP) that can be modified postpolymerization to allow for chemical cross-linking of the TGMs in situ, and incorporation of nonreactive hydrophilic side groups (AAm) to elevate the TGM LCST to allow for soluble degradation products at physiologic temperature. To this end, statistical copolymers of various compositions were synthesized from the monomers NiPAAm, MAEP, and AAm via AIBN-initiated free radical polymerization in DMSO (Scheme 1), resulting in TGMs with LCSTs above physiologic temperatures (Table 3) in 85–95% yields. Initial experiments found DMSO to be a more suitable solvent than less polar solvents, such as dioxane and tetrahydrofuran, for synthesis of the TGMs, as, once formed, the copolymer was not soluble in these solvents and readily precipitated out of solution (data not shown). The protocol outlined in the Materials and Methods sections resulted in copolymers that remained in DMSO solution. 1HNMR spectra indicated copolymers were formed with monomer ratios similar to feed ratios, as shown in Table 3. The copolymers had Mn ranging from 22 to 24 kDa and PDIs from 3.7 to 4.0, as determined by SEC.
Table 3. Composition and Lower Critical Solution Temperature (LCST) Characterization of Various Thermogelling Macromers before and after Esterification.
| monomer feed (NiPAAm/MAEP/AAm) | experimental feeda (NiPAAm/MAEP/AAm) | LCSTb | GMA mol%a | modified LCSTb |
|---|---|---|---|---|
| 74/8/18 | 74.3/7.5/18.2 | 51.8 ± 0.6 | 8.4 | 36.6 ± 0.2 |
| 80/8/12 | 79.3/8.7/12.0 | 43.9 ± 0.6 | 8.9 | 33.5 ± 0.1 |
| 70/12/18 | 71.4/11.6/17.0 | 53.1 ± 0.3 | 11.5 | 35.5 ± 0.4 |
| 76/12/12 | 75.6/11.8/12.6 | 46.1 ± 0.4 | 11.3 | 31.8 ± 0.2 |
| 75.5/10/14.5c | 74.6/9.8/15.6 | 48.7 ± 0.2 | 9.4 | 34.0 ± 0.1 |
| 72.5/13/14.5c | 71.6/12.9/15.5 | 49.7 ± 0.5 | 12.6 | 30.2 ± 0.4 |
Determined by 1H nuclear magnetic resonance spectroscopy
Determined by differential scanning calorimetry (n = 3)
Formulation selected for use in hydrogel characterization experiments
A full factorial design was used to evaluate the effect of MAEP and AAm on LCST of the TGMs, with values shown in Tables 1 and 2. As shown in Figure 2, main effects analysis revealed that an increase in MAEP from 8 to 12 mol % resulted in an increase in LCST of 0.21 °C for every 1 mol % MAEP substituted for NiPAAm and that an increase in AAm from 12 to 18 mol % resulted in an increase of 0.62 °C for every 1 mol % AAm substituted for NiPAAm. The interaction of the MAEP and AAm on LCST was not significant (p = 0.15). Additionally, the two TGMs selected for hydrogel characterization experiments underwent catalytic degradation with ALP, resulting in a significant decrease in LCST, as shown in Figure 3.
Figure 2.

Main effects of monoacryloxyethyl phosphate (MAEP) and acrylamide (AAm) incorporation, as well as their interaction (AAmxMAEP) on thermogelling macromer lower critical solution temperature (LCST). A positive number indicates that the particular parameter had an increasing effect on the LCST as it was changed from a low level (−) to a high level (+) as described in Table 2; * indicates statistical significance (p < 0.05). Error bars show standard error of the effect (n = 3).
Figure 3.

Modulation of lower critical solution temperature (LCST) of TGMs with 10 and 13 mol % monoacryloxyethyl phosphate (MAEP) selected for use in hydrogel characterization. Bars that share letters are not statistically different from one another (p > 0.05). Error bars show standard deviation (n = 3).
MA-TGM Synthesis and Characterization
The primary design criterion for the composition of the MA-TGMs was the attachment of hydrophobic cross-linkable groups that serve the dual purpose of decreasing the LCST and allowing for chemical cross-linking of the MA-TGM chains. The P–OH groups of phosphates in small molecules have been shown to be esterified via reaction with epoxide groups.17,18 The reaction conditions were modified to attach hydrophobic, chemically cross-linkable methacrylate groups to the TGM backbones described above via ring-opening phosphate esterification of GMA. 1H NMR spectra indicated that ester bonds connected to cross-linkable methacrylate groups replaced approximately 50% of available P–OH groups after the esterification described in Scheme 2. As shown in Table 1, LCSTs decreased with increasing GMA incorporation. TGMs with lower feeds of AAm resulted in smaller changes in LCST despite having similar GMA content as measured by NMR.
Two copolymer formulations with molar feeds of 10 and 13 mol % MAEP and 14.5 mol % AAm were selected for use in hydrogel characterization. These feeds were selected so that the TGMs would form dual-gelling hydrogels at physiologic temperature following esterification and become soluble at physiologic temperature after removal of the phosphate groups via degradation, as shown in Figure 3. While the pre-esterification and postdegradation LCSTs were not statistically different between the two groups, the esterified 13% MAEP formulation had higher GMA incorporation as expected, resulting in a significantly lower LCST than the 10% MAEP formulation.
Hydrogel Characterization
In order to investigate the hypothesized potential of chemical cross-linking to mitigate hydrogel syneresis, hydrogel swelling ratios of the two selected MA-TGM formulations, with and without APS/TEMED initiated chemical cross-linking, were evaluated at formation and after 24 h in PBS. Hydrogels that were not chemically cross-linked underwent visible syneresis (images not shown) during formation in the molds, while those that were cross-linked maintained the shape of the mold. Figure 4 shows representative examples of hydrogels formed via thermogelation at 37 °C as well as chemical gelation at ambient temperature. These images demonstrate thermogelation occurs in under a minute at 37 °C, while the first signs of chemical gelation do not occur until 10 min after radical initiation.
Figure 4.
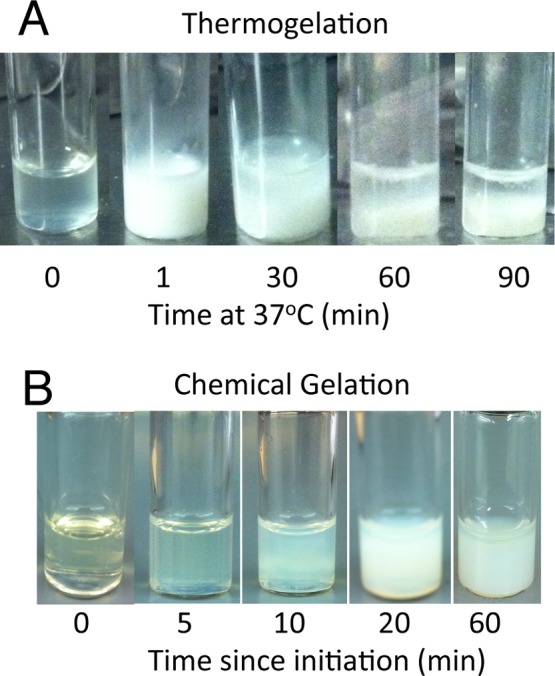
Representative images of methacrylated thermogelling macromers undergoing (A) thermogelation and subsequent phase separation (syneresis) at 37 °C and (B) chemical gelation at ambient temperature.
Quantitatively, Figure 5 shows that chemically cross-linked gels had significantly higher swelling ratios than the gels that were not chemically cross-linked at all time points. The 13% MAEP hydrogels that were not chemically cross-linked had significantly lower swelling ratios than the 10% MAEP gels that were not chemically cross-linked, both at formation and after 24 h in PBS. Furthermore, it should be noted that dual-gelled 10% MAEP hydrogels had significantly higher swelling ratios at 24 h than at formation, while 13% MAEP hydrogels did not undergo significant changes in swelling ratio during this time frame.
Figure 5.

Swelling ratio of hydrogels made from macromers with 10 and 13 mol % monoacryloxyethyl phosphate (MAEP) with and without chemical cross-linking. Bars that share letters are not statistically different from one another (p > 0.05). Error bars show standard deviation (n = 5).
To assess hydrogel degradation behavior, normalized hydrogel weight was monitored in PBS with or without ALP (200U/mL). As the phosphate ester bonds degrade, they are replaced with alcohols and phosphoric acid groups, leading to increased macromer hydrophilicity as well as a decrease in chain cross-links stabilizing the gel. Consequently, all groups demonstrated an increase in normalized weight over the course of the study secondary to hydrogel swelling, as shown in Figure 6. After 28 days in PBS, the presence of ALP resulted in significant increases in normalized weight compared to identical formulations incubated without ALP. Furthermore, the 10% MAEP hydrogels incubated without ALP had significantly larger normalized weight than 13% MAEP hydrogels incubated without ALP.
Figure 6.

Degradation of hydrogels composed of 10 and 13 mol % monoacryloxyethyl phosphate (MAEP) in the presence and absence of alkaline phosphatase (ALP); * indicates formulations incubated with ALP have significantly higher normalized weight than identical formulations incubated without ALP (p < 0.05). Error bars show standard deviation (n = 3).
FTIR spectroscopy was used to confirm that the phosphate ester bonds were being hydrolyzed. Figure 7 shows dramatic increases in peaks at 1043 and 3290 cm–1 with degradation, as indicated by increased normalized weight in both the 10 and 13% MAEP hydrogels. Respectively, these peaks correspond to the C–O stretch and the O–H stretch of primary alcohols, which are generated by phosphate ester hydrolysis.
Figure 7.

FTIR spectra of hydrogels composed of (A) 10 and (B) 13 mol % monoacryloxyethyl phosphate (MAEP) after 1 day in PBS and after 28 days in PBS with or without alkaline phosphatase (ALP). Peaks at 1043 cm–1 (x) and 3090 cm–1 (y) correspond to the C–O stretch and the O–H stretch, respectively, of primary alcohols generated by phosphate ester hydrolysis and increase with increased degradation.
In an effort to determine the hydrogels’ potential for use in bone regeneration, calcium deposition within the hydrogels over time was measured. Significant increase in calcium deposition was not noted in any of the gels until day 15 (Figure 8). Interestingly, the 10% MAEP had higher levels of bound calcium than MAEP = 13% at both day 15 and day 20 time points, despite having less total phosphorus content.
Figure 8.

Calcium content of hydrogels composed of 10 and 13 mol % monoacryloxyethyl phosphate (MAEP) following incubation in complete osteogenic medium. Bars that share letters are not statistically different from one another (p > 0.05). Error bars show standard deviation (n = 4).
The cytocompatibility of the hydrogels was evaluated by fluorescent Live/Dead analysis of rat fibroblasts after 2 and 24 h of exposure to hydrogel leachables. As seen in Figure 9A, at the 2 h time point, no difference was seen between the live control and any of the experimental groups. At the 24 h time point (Figure 9B), the 1× 10% MAEP group had significantly lower percentage of live cells than the live control. All other groups were not statistically different from the live control.
Figure 9.

Cytotoxicity of leachables of hydrogels composed of 10 and 13 mol % monoacryloxyethyl phosphate (MAEP) at (A) 2 and (B) 24 h. Columns that share the same letter are not statistically different (p > 0.05). Error bars show standard deviation (n = 6).
Discussion
TGM and MA-TGM Characterization
TGMs were successfully synthesized with calculated monomer ratios, similar to the feed ratios. The NiPAAm incorporation was sufficient to yield thermoresponsive copolymers. The appearance of a single peak while measuring LCST, both before and after esterification, suggests the monomers were evenly distributed on the TGM backbone. The factorial study evaluated with DSC was used to elucidate the effect of increasing AAm and MAEP molar feeds on TGM LCST and demonstrate that the LCST of the TGMs can be predictably tuned by varying monomer molar feed. Furthermore, degraded TGM LCST could be evaluated by hydrolyzing the phosphate ester bonds with alkaline phosphatase at 37 °C, resulting in a significant decrease in LCST, as seen in Figure 3. This drop was expected, as the phosphate group was removed from the TGM and replaced with a less hydrophilic primary alcohol. Nevertheless, these degraded TGM LCSTs remained well above physiologic temperature. MA-TGMs were synthesized via esterification of the phosphate groups of MAEP by GMA, with incorporation confirmed by 1H NMR. The amount of GMA incorporated was dependent on the amount of phosphate groups available. However, the magnitude of the resulting decrease in LCST was dependent on both the amount of GMA incorporated and the initial LCST of the TGM. TGMs with higher initial LCSTs had larger drops in LCST following esterification, despite similar GMA incorporation, indicating that the attachment of hydrophobic groups has a lesser effect on decreasing LCST with increasing hydrophobicity of the initial TGM. This information was used to select the two MA-TGM formulations that were used for hydrogel characterization.
Finally, it should be noted that the Mn of these NiPAAm-based TGMs are similar to other NiPAAm-based TGMs that have been shown to undergo rapid glomerular filtration,19 making them promising candidates for future use in vivo. Moreover, the large PDIs are likely due to impurities in the MAEP monomer, which has been shown to contain varying amounts of diacrylated phosphates,20 leading to branched copolymers connected via degradable phosphate ester bonds.
Hydrogel Characterization
Two MA-TGM formulations were selected for hydrogel characterization based on their ability to form stable, dual-cross-linked hydrogels at physiologic temperature and have soluble degradation products, making them promising candidates for in vivo applications. Both of these formulations had significantly lower swelling ratios when they did not undergo chemical cross-linking, indicating that chemical cross-links can mitigate the syneresis of the hydrogels. This can be visualized in Figure 4, which demonstrates the primary initial gelation mechanism is thermogelation. Moreover, the 10% MAEP hydrogels underwent significant swelling in the first 24 h, while the 13% MAEP hydrogels did not significantly change in that time frame, though it did trend upward. This upward trend in swelling ratio is likely due to a small increase in hydrophilicity as the methacrylate groups are cross-linked to form a saturated carbon chain. Furthermore, the chemically cross-linked 10% MAEP hydrogels likely had a larger increase in swelling ratio than the chemically cross-linked 13% MAEP hydrogels after 24 h in PBS because of the larger number of chemically cross-linkable groups available in the 13% MAEP formulation, yielding a more cross-linked, less flexible copolymer network. Though not statistically significant, the formulations that were not chemically cross-linked demonstrated the opposite trend, decreased swelling ratio after 24 h in PBS, as is common in thermogelling polymers that are not chemically cross-linked.
The hypothesis that hydrogels made from 13% MAEP formulation form a more cross-linked, less flexible network is also supported by the degradation study. The slowed rate of swelling in 13% MAEP hydrogels indicates degradation of the hydrogels can be modified by varying the number of chemically cross-linkable GMA groups present at hydrogel formation. Additionally, the degradation study showed that ALP accelerates the hydrolysis of the phosphate ester bonds of the hydrogel. This can be favorable for bone tissue engineering applications, as ALP-producing bone cells infiltrating or differentiating within the hydrogel can accelerate the degradation rate locally and possibly allow for improved cellular migration and proliferation in these areas.
The hydrogel mineralization data suggest that the higher cross-linking density of the 13% MAEP hydrogels slows the diffusion of molecules in and out of the hydrogel. Significant increase in calcium bound to the hydrogels was not detectable until day 15. A possible cause for the delay in detectable calcium is that the phosphorus nucleation sites should increase with time, secondary to phosphate ester degradation. Additionally, as cross-links degrade, serum proteins present in complete osteogenic media can diffuse into the gel and facilitate mineralization. At days 15 and 20, the 10% MAEP hydrogels had significantly more calcium than the 13% MAEP hydrogels, despite having less overall phosphorus content. The most likely cause for the 10% MAEP hydrogels to have more bound calcium is that the relatively less cross-linked copolymer network results in higher diffusion coefficients in the hydrogel when compared to 13% MAEP hydrogels. This suggests that a major driving force in hydrogel mineralization is the diffusion of larger molecules such as serum proteins into the hydrogel. This hypothesis is further supported by the hydrogel leachable cytotoxicity data also seems to indicate that the 13% MAEP hydrogels are heavily cross-linked enough to provide a decreased diffusion coefficient to cytotoxic molecules. The only group that had a significantly lower value than the live control was the 10% MAEP hydrogels at 24 h of exposure. While some cytotoxicity is to be expected when using APS/TEMED-initiated systems, why only the 10% MAEP formulation had a lower percentage of live cells than the control is not clear. However, this could be explained by the incomplete diffusion of cytotoxic leachables, such as the APS and TEMED, from the 13% MAEP hydrogels due to a smaller diffusion coefficient, resulting in hydrogel-conditioned media containing less cytotoxic leachables than the 10% MAEP hydrogel-conditioned media. Summarily, the 10% MAEP hydrogels appear to have a higher diffusion coefficient due to relatively decreased cross-linking density, which could make it more fit for cell-delivery applications than the MAEP-13% hydrogels.
Conclusions
A novel, thermogelling, p(NiPAAm)-based macromer with pendant phosphate groups was synthesized and subsequently functionalized with chemically cross-linkable methacrylate groups via degradable phosphate ester bonds, yielding an injectable, degradable dual-gelling macromer. The relationship between monomer feed concentration and LCST was elucidated, allowing the LCST of the TGM to be tuned for in situ gelation at physiologic temperature while maintaining soluble degradation products. Additionally, the dual gelation mitigated hydrogel syneresis, making this a promising material for defect-filling, cellular encapsulation applications. Finally, the ability of these phosphorus-containing hydrogels to mineralize in vitro warrants further investigation as a bone tissue engineering material.
We acknowledge support by the National Institutes of Health (R01 DE17441 and R01 AR48756), the Keck Center Nanobiology Training Program of the Gulf Coast Consortia (NIH Grant No. T32 EB009379), and the Baylor College of Medicine Medical Scientist Training Program (NIH T32 GM007330).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Salinas C. N.; Anseth K. S. J. Dent. Res. 2009, 88, 681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretlow J. D.; Young S.; Klouda L.; Wong M.; Mikos A. G. Adv. Mater. 2009, 21, 3368–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klouda L.; Mikos A. G. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild H. G. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar]
- Hacker M. C.; Klouda L.; Ma B. B.; Kretlow J. D.; Mikos A. G. Biomacromolecules 2008, 9, 1558–1570. [DOI] [PubMed] [Google Scholar]
- Ekenseair A. K.; Boere K. W. M.; Tzouanas S. N.; Vo T. N.; Kasper F. K.; Mikos A. G. Biomacromolecules 2012, 13, 1908–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klouda L.; Perkins K. R.; Watson B. M.; Hacker M. C.; Bryant S. J.; Raphael R. M.; Kasper F. K.; Mikos A. G. Acta Biomater. 2011, 7, 1460–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z.; Lee B. H.; Vernon B. L. Biomacromolecules 2007, 8, 1280–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J.; Hong Y.; Ma Z.; Wagner W. R. Biomacromolecules 2008, 9, 1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet D. J.; Dhruv H. D.; Vernon B. L. Biomacromolecules 2010, 11, 1154–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millán J. L. Purinergic Signalling 2006, 2, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.-A.; Williams C. G.; Yang F.; Cher N.; Lee H.; Elisseeff J. H. Tissue Eng. 2005, 11, 201–213. [DOI] [PubMed] [Google Scholar]
- Nuttelman C. R.; Benoit D. S. W.; Tripodi M. C.; Anseth K. S. Biomaterials 2006, 27, 1377–1386. [DOI] [PubMed] [Google Scholar]
- Chirila T. V.; Zainuddin; Hill D. J. T.; Whittaker A. K.; Kemp A. Acta Biomater. 2007, 3, 95–102. [DOI] [PubMed] [Google Scholar]
- Henslee A. M.; Gwak D.-H.; Mikos A. G.; Kasper F. K. J. Biomed. Mater. Res., Part A 2012, 100, 2252–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmer M. D.; Shin H.; Horch R. A.; Ambrose C. G.; Mikos A. G. Biomacromolecules 2003, 4, 1026–1033. [DOI] [PubMed] [Google Scholar]
- Osanai S.; Yamada G.; Hidano R.; Beppu K.; Namiwa K. J. Surfactants Deterg. 2009, 13, 41–49. [Google Scholar]
- Tuzhikov O. I.; Khokhlova T. V.; Bondarenko S. N.; Dkhaibe M.; Orlova S. a. Russ. J. Appl. Chem. 2009, 82, 2034–2040. [Google Scholar]
- Bertrand N.; Fleischer J. G.; Wasan K. M.; Leroux J.-C. Biomaterials 2009, 30, 2598–2605. [DOI] [PubMed] [Google Scholar]
- Grøndahl L.; Suzuki S.; Wentrup-Byrne E. Chem. Commun. (Cambridge, U. K.) 2008, 3314–3316. [DOI] [PubMed] [Google Scholar]