

Abstract
The potential therapeutic benefit of compounds able to activate AMPA receptors (AMPAr) has led to the search for new AMPAr positive modulators. On the basis of crystallographic data of the benzothiadiazines binding mode in the S1S2 GluA2 dimer interface, a set of 5-aryl-2,3-dihydrobenzothiadiazine type compounds has been synthesized and tested. Electrophysiological results suggested that 5-heteroaryl substituents on the benzothiadiazine core like 3-furanyl and 3-thiophenyl dramatically enhance the activity as positive modulators of AMPAr with respect to IDRA21 and cyclothiazide. Mouse brain microdialysis studies have suggested that 7-chloro-5-(3-furyl)-3-methyl-3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxide crosses the blood–brain barrier after intraperitoneal injection. Biological results have been rationalized by a computational docking simulation that it has currently employed to design new AMPAr-positive modulator candidates.
Keywords: AMPA modulators, IDRA21, benzothiadiazines, cognitive enhancers
The principal excitatory neurotransmitter in the mammalian central nervous system (CNS) is l-glutamate, and its signal transduction is mediated by ionotropic and metabotropic receptors.1 Different studies suggest that ionotropic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAr) is involved in learning processes and in memory establishment.2 The therapeutic potential of compounds able to activate AMPAr has led to the search for new AMPAr-positive modulators.3,4 Among these, one of the most investigated chemical classes of compounds are benzothiadiazine derivatives such as cyclothiadiazide (CTZ) (1), (±)7-chloro-3-methyl-3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxide (IDRA21) (2), and (±) 8-chloro-2,3,5,6-tetrahydro-3,6-dimethyl-pyrrolo[1,2,3-de]-1,2,4-benzothiadiazine 1,1-dioxide (3), which are able to inhibit desensitization of AMPAr-potentiating ionotropic glutamatergic neurotransmission (Figure 1).5−7
Figure 1.

Cyclothiazide (1), IDRA21(2), and 8-chloro-2,3,5,6-tetrahydro-3,6-dimethyl-pyrrolo[1,2,3-de]-1,2,4-benzothiadiazine 1,1-dioxide (3).
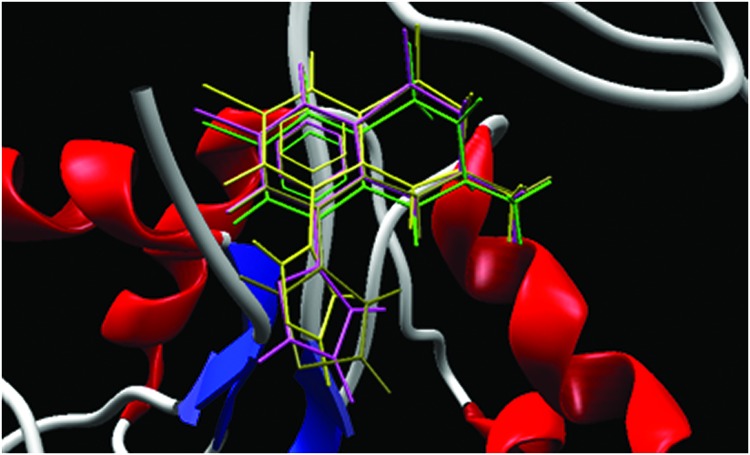
IDRA21, the first benzothiadiazine effective in increasing learning and memory performance in behavioral tests, represents an important lead compound since it is able to cross the blood–brain barrier (BBB).8,9 Recent crystallographic studies regarding the binding mode of benzothiadiazine derivatives on S1S2 GluA2 subunits of AMPAr suggest that these compounds bind to the dimer interface, leading to inhibition of the receptor desensitization by stabilization of the ligand-binding domain within a dimer interface.10,11 Notwithstanding structure similarities of IDRA21 and cyclothiazide, crystal structures of their complexes with S1S2 GluA2 domain reveal different binding modes.10,11 In particular, IDRA21 and cyclothiazide bind to the dimer interface S1S2 GluA2 with a different orientation probably due to lower steric hindrance of C3 methyl group of IDRA21 with respect to the C3 norbornyl substituent of cyclothiazide (Figure 2).10,11
Figure 2.
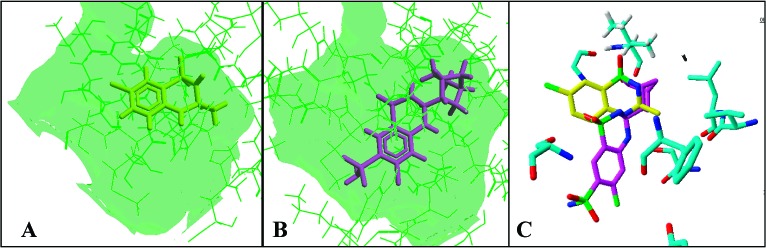
Comparison of binding modes of IDRA21 (A) (PDB code: 3IL1) and cyclothiazide (B) (PDB code: 1LBC) in the ligand binding site of S1S2 GluA2 dimer interface. Overlap view of IDRA21 and cyclothiazide (C).
The crystal structure of cyclothiazide in complex with S1S2 GluA2 subunits (PDB code: 1LBC) reveals that the benzene ring and C7 sulfonamidic group of cyclothiazide fit into a hydrophilic pocket defined by Lys763, Tyr424, Ser497, Phe495 and Ser729 residues. In contrast, the same hydrophilic pocket is unfilled in the crystal structure of IDRA21 in complex with S1S2 GluA2 subunits.
Detailed analysis of the latter crystal structure reveals that the pocket contains four water molecules ordered to form a network of hydrogen bonds with neighboring residues. One of these water molecules is positioned on the plane of IDRA21 aromatic ring at a distance of 3.2 Å from the C5 atom. Therefore, we hypothesized that the affinity of IDRA21 derivatives toward AMPAr could be increased by a substituent at C5 position of IDRA21 core that could accommodate the empty hydrophilic pocket and form additional hydrogen bonds with the receptor and water molecules present in this cavity.
Molecular modeling studies indicate that heteroaromatic substituents, like thiophenyl or furanyl, and aromatic substituents like anilinic or benzoyl on C5 position of IDRA21 can fit into the unfilled hydrophilic pocket, miming the binding mode of benzene ring and C7 sulfonamidic group of cyclothiazide (Figure 3).12
Figure 3.
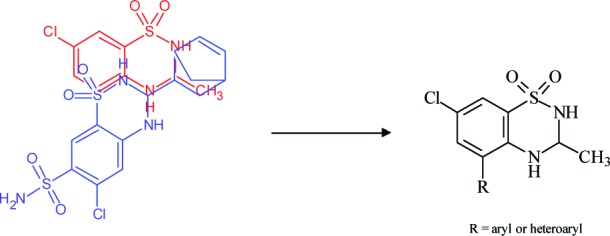
Overlay of IDRA21 (red) and cyclothiazide (blue) led to the design of new IDRA21 analogues.
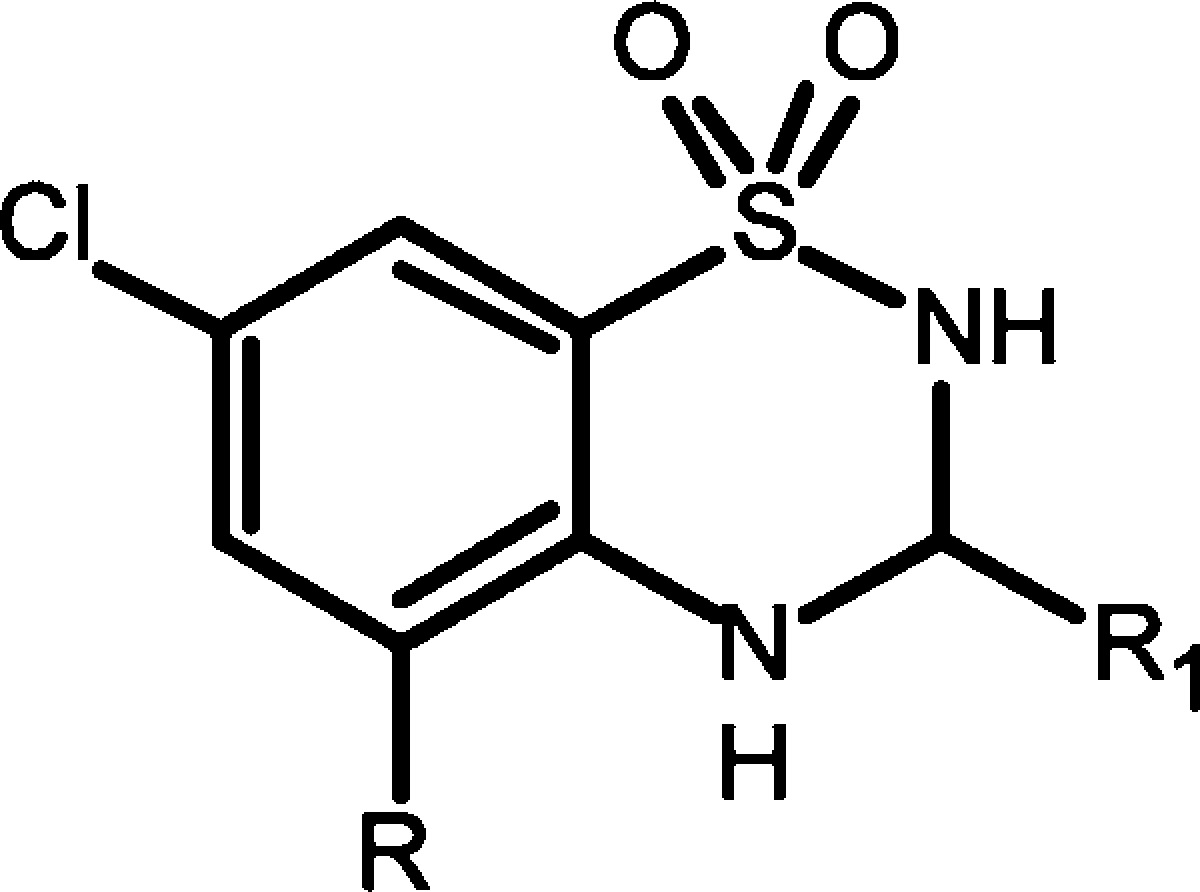
Hence, several 5-aryl-7-chloro-3-methyl-3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxide derivatives were prepared by a synthetic route based on the Pd-catalyzed Suzuki–Miyaura coupling reaction (Table 1). The general synthetic pathway employed is shown in Scheme 1.
Table 1. Potentiation of Kainate (100 μM)-Evoked Current Induced by the Synthesized Compound at 10 μM.
Each value is the mean ± SEM of 5–8 experiments.
Scheme 1.

Halogenation of 2-amino-5-chlorobenzensulfonamide (4a) in acidic conditions gave 4b, which subsequently afforded intermediate 4c by Pd-catalyzed cross-coupling reaction with a suitable boronic acid. The 5-aryl-7-chloro-3-methyl-3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxide derivatives were obtained by ring closure with a selected aldehyde under catalytic acidic conditions. Compounds 11 and 12 were obtained by an alternative synthetic pathway: The coupling reaction with 3-aminophenylboronic acid was conducted on 7-chloro-5-iodo-3-alkyl-3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxide, since aldehyde reacted with the nitrogen atom of the anilinic group of the aromatic substituents at C5 during benzothiadiazine ring closure (Scheme 2).
Scheme 2.

The prepared compounds described in Table 1, as well as their intermediates, were fully characterized by IR, ESI, and NMR spectroscopic analysis. Subsequently, the activities of synthesized compounds were evaluated by patch-clamp technique in primary cultures of cortical neurons as allosteric modulators of AMPA/kainate receptors.13,14 The capability of potentiating kainate-evoked current was tested for all of the newly synthesized compounds. Kainate-evoked current was mainly mediated by AMPA receptor activation because application of GYKI 53655 (100 μM), an AMPA receptors antagonist, almost completely abolished the current (data not shown). Because 3 is one of the most active benzothiadiazine derivatives reported in literature as a AMPAr-positive allosteric modulator, it was selected as reference compound.4 Data obtained indicate that 5, 9, and 10, at 10 μM, potentiate by 80 ± 16, 358 ± 80, and 214 ± 41%, respectively, the kainate-evoked currents at 10 μM (Tab.1). The dose–response curve of the most active compound 9 reveals an EC50 of 1.3 μM, suggesting that this benzothiadiazine derivative is one of the most potent within this class of AMPAr-positive allosteric modulators (Figure 4).
Figure 4.
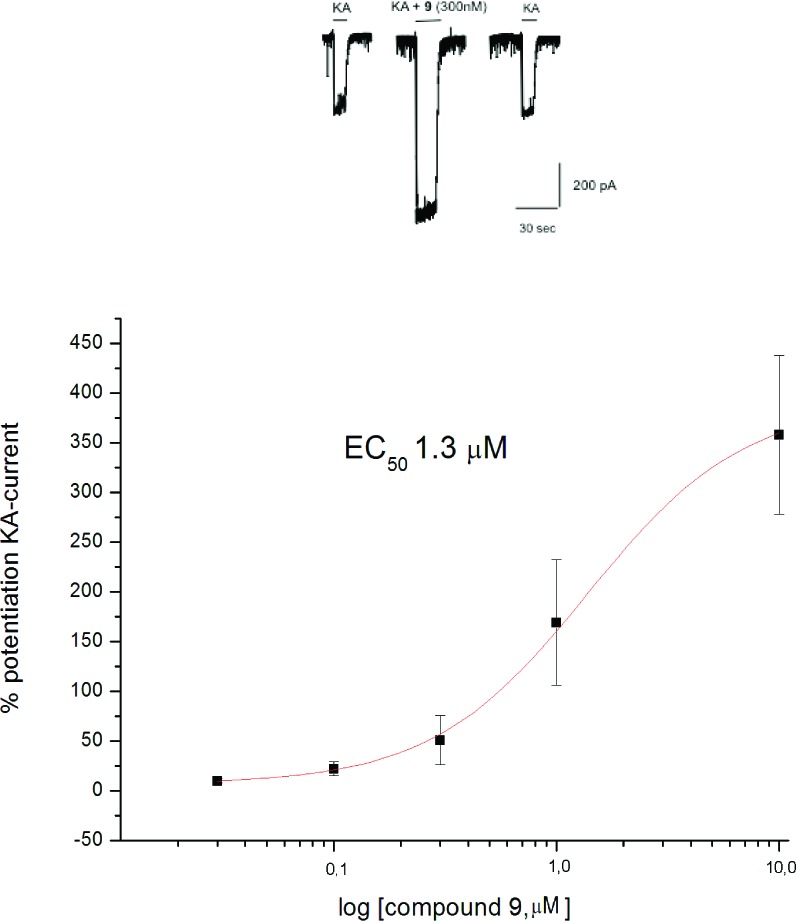
Modulation of KA-evoked current by compound 9. (a) Representative whole cell recording showing KA (100 μM)-evoked current recorded from a cortical neuron in culture in control conditions (left traces), after application of 9 (300 nM) (middle trace) and after washing (right trace). (b) Dose–response curve of the effect of 9 on KA-evoked current. Each data point is the mean ± SEM of at least five cells.
The analysis of these preliminary biological results suggests a net of different molecular effects of these ligands, which affects the potentiation of KA-current activity. In particular, the activity significantly increases with a five-membered aromatic ring as the C5 substituent, whereas compounds with a six-membered aromatic ring show a very low activity. This effect is probably due to a sterical hindrance that prevent them from fitting into the hydrophilic pocket at S1S2 GluA2 dimer interface. Moreover, the maximum of activity is achieved when the heteroatom is at 3-position of the heteroaryl substituent. The activities of compounds 5–7 indicate that the C3 substituent has a critical influence . The preferred substituent appears to be CH3 (5) since H or CH2CH3 led to compounds (6 and 7) with lower activity as KA-current potentiators. Because of its activity, 9 represents a promising lead compound for the development of new potent AMPAr-positive allosteric modulators.
Molegro Virtual Docker (MVD) software has been employed to dock 5, 9, and 10 into the ligand binding site of S1S2 GluA2 dimer (PDB code: 3IL1) to understand their binding mode.12 The software MVD was evaluated on cyclothiazide, triflumethiazide, diazoxide, althiazide, hydrochlorothiazide, chlorothiazide, trichlormethiazide, and IDRA21. The average root-mean-square distance (rmsd) of the best ranking pose of tested compounds as compared to their binding pose in the respective crystal structures was found to be 0.81 Å, proving that MVD is able to accurately dock this type of compounds (see the Supporting Information). Compounds 5, 9, and 10 compounds were built by the software Spartan'08 and energy minimized using Hartree–Fock ab initio method employing at the SCF level with the 6-31G* basis set.
The docking output clearly indicates that 5, 9, and 10 bind similarly to IDRA21 at the dimer interface of S1S2 GluA2 subunits with the C3 methyl substituent placed into the hydrophobic cleft defined by Val750, Leu751, and Leu259 residues, the hydrogen atom on N4 of benzothiadiazine gave an hydrogen bond with Ser754 (H-bond length 3.39 Å) residue, and the sulfone oxygen atom of benzothiadiazine gave a polar interaction with Gly731 residue (Figure 5) since it is located within 2.78 Å from the backbone of this residue. These interactions are observed also in the crystal structure of IDRA21 in complex with S1S2 GluA2 subunits.
Figure 5.
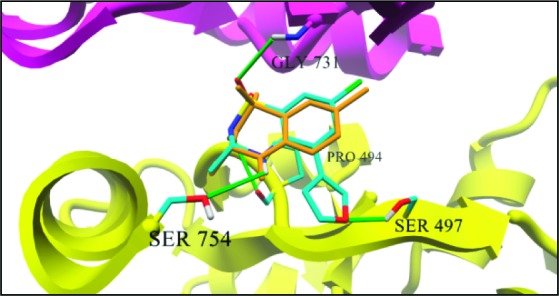
Binding of 9 and IDRA21 to the S1S2 GluA2 dimer interface; the secondary structures of the two S1S2 monomers are colored in yellow and magenta; green sticks illustrate plausible hydrogen bond interactions between 9 and the receptor residues, both colored in element color code; the IDRA21 molecule present in the crystal model is colored in orange.
The increased activity of 5, 9, and 10 with respect to IDRA21 can be explained by additional interactions with the ligand binding site due to heteroaryl substituent in the C5 position of the benzothiadiazine core that can be accommodated in the unfilled hydrophilic pocket previously recognized in the crystal structure of S1S2 GluA2 domain in complex with IDRA21.
Compounds 9 and 10 have shown higher activity with respect to 5 probably because of a H-bond between the heteroatom of 3-furanyl or 3-thiophenyl substituent with the Ser497 residue of S1S2 GluA2 domain. In particular, the heteroatom of the C5 substituent of 9 and 10 is within hydrogen bond distance (2.71 Å) to the hydroxyl group of Ser497. Moreover, 9 is more active than 10 probably due to the major electronegativity of oxygen atom of 3-furanyl that allows a stronger H-bond with respect to the sulfur atom of the 3-thiophenyl substituent.
The good activity of 9 as positive allosteric modulator of AMPAr in vitro suggests that it could exert cognition enhancing properties when administered in vivo. With the aim to evaluate if 9 can elicit AMPAr activity on the CNS, its capability to cross the BBB was investigated. Recently, it has been reported that 3,4-dihydro-2H-1,2,4-benzothiadiazine derivatives in vivo can hydrolyze in acidic conditions to corresponding 2-aminobenzensulfonamide.15 Moreover, Pirrotte et al. have recently reported that 4-alkyl-3,4-dihydro-2H-1,2,4-benzothiadiazine derivatives undergo the action of hepatic cytochrome P450, giving the corresponding 4-alkyl-2H-1,2,4-benzothiadiazine derivative.16 As a consequence, it is possible that 9 in vivo can be hydrolyzed to 5-chloro-3-(3-furyl)-2-aminobenzensulfonamide (16) and metabolized to give 7-chloro-5-(3-furyl)-3-methyl-4H-1,2,4-benzothiadiazine 1,1-dioxide (17).
In the attempt to evaluate if 9 crosses the BBB and if it produces 16 and 17 in vivo, cerebral microdialysis experiments have been performed in mice to measure cerebral levels of 9, 16, and 17 after intraperitoneal administration of 9. A liquid chromatography tandem mass spectrometry (LC/MS/MS) method has been developed to evaluate concentrations of parent ompound 9 and of their possible metabolites 16 and 17 in cerebral mouse dialysates. The identity of the compounds 9, 16, and 17 was confirmed by comparing their retention times and their MS/MS profiles after injection of the corresponding synthetic authentic compounds in the HPLC system. Figure 6 shows the extracellular levels of 9 and 17 in mouse nucleus accubens after intraperitoneal administration of 9 (20 mg/kg).
Figure 6.

Time course of 9 and 17 outputs from mouse nucleus accumbens after a single administration of 9 (20 mg/kg ip). Values are expressed as μM of mean values ± SEMs of dialysates concentrations.
Dialysate concentrations of 16 were under the limit of detection of the analytical method and are not reported in the graph. The concentrations of 9 increased in the first 40 min after administration to reach the concentration of 37 μM and then gradually decreased and became undetectable after 2 h. Levels of 17 increased in the first 1 h to reach the concentration of 8 μM, then decreased slowly for the next 2 h to be undetectable at 140 min. The microdialysis results indicate that 9 reaches the CNS after intraperitoneal injection. Moreover, 9 is subject to a moderate action of cytocrome P450 to give 17, while it does not undergo hydrolysis after intraperitoneal administration. In summary, a novel class of positive allosteric modulators of AMPAr has been designed based on crystal structures of IDRA21 and cyclothiazide in complex with the S1S2 GluA2 dimer interface. An efficient and versatile synthetic route has been developed to prepare the designed 5-aryl-3,4-dihydrobenzothiadiazine compounds. Electrophysiological results suggested that 5-heteroaryl substituents like 3-furanyl and 3-thiophenyl dramatically enhance the activity as positive modulators of AMPAr with respect to IDRA21 and cyclothiazide. Biological results have been rationalized by a computational docking simulations that are employed to design new AMPAr-positive modulator candidates. Moreover, mouse cerebral microdialysis studies suggested that 9 crosses the BBB after intraperitoneal injection since it has been detected at a micromolar concentration in mouse nucleus accumbens dialysates. Low levels of the corresponding unsatured 17 derivative have been detected in mouse nucleus accumbens dialysates, suggesting a moderate action of cytocrome P450 on 9. Efforts aimed at a further optimization, as well as in-depth biological studies, of the novel compounds are ongoing, and the results will be reported in due course.
Acknowledgments
We thank Prof. M. Zoli and Dr. S. Guiducci for the help in microdialysis experiments.
Supporting Information Available
Experimental procedures, compound characterizations, computational methods, and pharmacology. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Dingledine R.; Borges K.; Bowie D.; Traynelis S. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [PubMed] [Google Scholar]
- Kew J. N. C.; Kemp J. A. Psychopharmacology 2005, 179, 4. [DOI] [PubMed] [Google Scholar]
- Black M. D. Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology 2005, 179, 154–163. [DOI] [PubMed] [Google Scholar]
- Swanson G. T. Targeting AMPA and kainate receptors in neurological disease: Therapies on the horizon?. Neuropsychopharmacology 2009, 34, 249–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L.; Patneau D. K.; Mayer M. L. Modulation of excitatorysynaptic transmission by drugs that reduce desensitization at AMPA/kainate receptors. Neuron 1991, 7, 971–984. [DOI] [PubMed] [Google Scholar]
- Bertolino M.; Baraldi M.; Parenti C.; Braghiroli D.; DiBella M.; Vicini S.; Costa E. Recept. Channels 1993, 1, 267. [PubMed] [Google Scholar]
- Philips D.; Sonnenberg J.; Arai A. C.; Vaswani R.; Krutzik P. O.; Kleisli T.; Kessler M.; Granger R.; Lynch G; Chamberline A. R. 5′-Alkyl-benzothiadiazides: A New Subgroup of AMPA Receptor Modulators with Improved Affinity. Bioorg. Med. Chem. 2002, 10, 1229–1248. [DOI] [PubMed] [Google Scholar]
- Arai A.; Guidotti A.; Costa E.; Lynch G. Effect of the AMPA receptor modulator IDRA 21 on LTP in hippocampal slices. NeuroReport 1996, 7, 2211–2215. [DOI] [PubMed] [Google Scholar]
- Thompson D. M.; Guidotti A.; DiBella M.; Costa E. 7-Chloro-3-methyl-3,4-dihydro-2H-1,2,4-benzothiadiazine S,S-dioxide (IDRA 21), a congener of aniracetam, potently abates pharmacologically induced cognitive impairments in patas monkeys. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 7667–7671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Olson R.; Horning M.; Armstrong N.; Mayer M.; Gouaux E. Mechanism of glutamate receptor desensitization. Nature 2002, 417, 245–253. [DOI] [PubMed] [Google Scholar]
- Ptak C. P.; Ahmed A. H.; Oswald R. E. Probing the Allosteric Binding Site of Glur2 with Thiazide derivatives. Biochemistry 2009, 48, 8594–8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R.; Christensen M. H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [DOI] [PubMed] [Google Scholar]
- Hamill O. P.; Marty A.; Neher E.; Sakmann B.; Sigworth F. J. Improved patch-clamp techniques for high-resolution currents recording from cells and cell-free membrane patches. Pflugers Arch. 1981, 391, 85–100. [DOI] [PubMed] [Google Scholar]
- Luo J. H.; Fu Z. Y.; Losi G; Kim B. G.; Prybylowski K; Vissel B; Vicini S. Functional expression of distinct NMDA channel subunits tagged with green fluorescent protein in hippocampal neurons in culture. Neuropharmacology 2002, 42, 306–318. [DOI] [PubMed] [Google Scholar]
- Cannazza G.; Jozwiak K.; Parenti C.; Braghiroli D.; Carrozzo M. M.; Puia G.; Losi G.; Baraldi M.; Lindner W.; Wainer I. W. A Novel class of allosteric modulators of AMPA/Kainate receptors. Bioorg. Med. Chem. Lett. 2009, 19, 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francotte P.; Goffin E.; Fraikin P.; Lestage P.; Van Heugen J. C.; Gillotin F.; Danober L.; Thomas J. Y.; Chiap P.; Caignard D. H.; Pirotte B.; de Tullio P. New Fluorinated 1,2,4-Benzothiadiazine 1,1-Dioxides: Discovery of an Orally Active Cognitive Enhancer Acting through Potentiation of the 2-Amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic Acid Receptors. J. Med. Chem. 2010, 53, 1700–1711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.