

Abstract
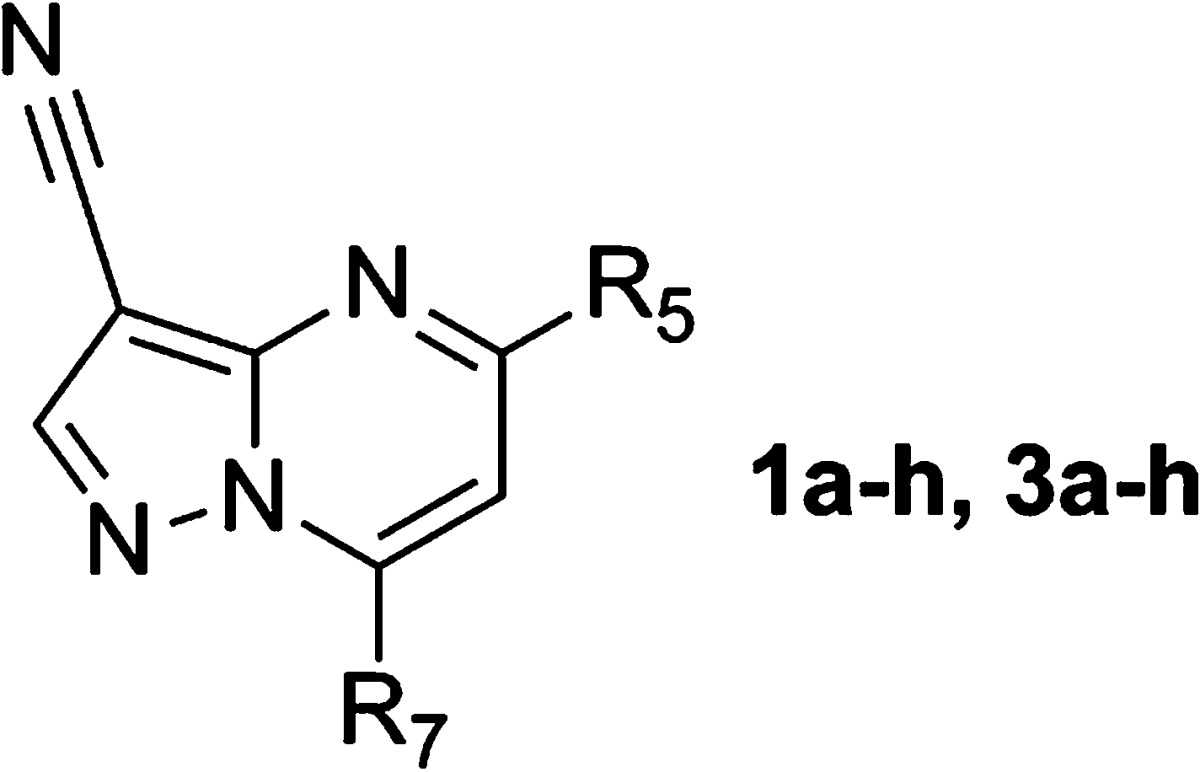
In this paper we describe a series of 3-cyano-5-aryl-7-aminopyrazolo[1,5-a]pyrimidine hits identified by kinase-focused subset screening as starting points for the structure-based design of conformationally constrained 6-acetamido-indole inhibitors of CK2. The synthesis, SAR, and effects of this novel series on Akt signaling and cell proliferation in vitro are described.
Keywords: CK2; casein kinase; structure-based design; pyrazolo[1,5-a]pyrimidine
Casein Kinase 2 (CK2) is a highly conserved and ubiquitously expressed serine/threonine kinase, which exists as a tetrameric complex containing two catalytic (α or α′) and two regulatory (β) subunits.1 CK2 is regarded as a constitutively active kinase with broad function, although more recent evidence suggests that CK2 can be regulated by growth factors such as IL-6 and epidermal growth factor (EGF).2,3 The importance of CK2 for cell viability has been demonstrated by genetic studies, showing that suppression of the α or β subunits leads to embryonic lethality in mice, while genetic silencing of the α′ subunit results in viable offspring, but with males exhibiting defects in spermatogenesis leading to infertility.4,5 CK2 regulates several key oncogenic signaling pathways, including PI3K (phosphatidylinositol 3-kinase), AKT (protein kinase B), NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells), and Wnt (wingless type MMTV integration site family).6−8 Targeted overexpression in transgenic animal models results in neoplastic growth9 and administration of antisense oligodeoxy nucleotides against CK2α induced concentration-dependent tumor shrinkage in a human prostate cancer xenograft model.10
A variety of ATP-competitive, small molecule inhibitors of CK2 have been identified.11 These include various polyhydroxylated aromatic compounds, such as emodin and quercetin, polyhalogenated compounds, such as DRB (5,6-dichlororibofuranosylbenzimidazole) and TBB (4,5,6,7-tetrabromo-1H-benzotriazole), as well as the indolo[1,2-a]quinazoline derivative IQA.12 More recently, researchers from Cylene have disclosed CX-4945, a selective, orally available small molecule inhibitor of CK2 which exhibits single-agent activity in xenograft models and is currently in clinical trials for the treatment of cancer.13
There remains a need for additional novel agents with the necessary balance of potency, selectivity, druglike properties, tolerability, and efficacy that can successfully exploit the critical function of CK2 in cancer-linked pathways for the treatment of disease. Herein we disclose the discovery of a series of potent and selective inhibitors of CK2 derived from the pyrazolo[1,5-a]pyrimidine scaffold.
As an alternative to conventional high throughput screening (HTS), we employed a kinase-focused subset screening approach. A library of ≈10K compounds was assembled from the AstraZeneca collection by applying general kinase binding-site and target-specific structural bioinformatic filters. Single point screening of the subset gave ≈2500 compounds with kinase inhibiton greater than 60% at 10 μM. These hits were consolidated further by application of physical property, in vitro ADME (absorption, distribution, metabolism, and excretion), and kinase selectivity filters to give ≈1400 compounds selected for full CK2 α′ IC50 determination using an in vitro mobility shift microfluidics assay.14 Among the hits characterized in this way, a series of 3-cyano-5-aryl-7-amino-pyrazolo[1,5-a]pyrimidines (Table 1, 1a–h) exhibited submicromolar potency and high ligand efficiency15 (1a; LE = 0.4). Preliminary SAR studies for 1a–h suggested that electron-rich aromatic and heteroaromatic systems at C5 and substitution of the C7 amine with small carbo- and heterocyclic substituents enhanced potency. Neither the des-cyano analogue of 1a nor the 2-methyl derivative of 1f (not shown) exhibited activity toward CK2 (>30 μM). However, despite the encouraging biochemical activity exhibited by this series, potency in Alamar Blue cell proliferation assays14 was modest (1g, HCT-116 GI50 = 4.5 μM).
Table 1. CK2 Biochemical and Cellular Potency for Compounds 1a–h and 3a–h.
| cpd | R5 | R7 | CK2 IC50 (μM)a | HCT-116 GI50 (μM)a |
|---|---|---|---|---|
| 1a | 2-thienyl | NH2 | 0.10 | >30 |
| 1b | Ph | NH2 | 0.18 | >30 |
| 1c | p-MeO-Ph | NH2 | 0.058 | ND |
| 1d | p-F-Ph | NH2 | 0.43 | ND |
| 1e | 2-thienyl | cyclopropylamino | 0.46 | ND |
| 1f | p-MeO-Ph | cyclopropylamino | 0.078 | 14 |
| 1g | p-MeO-Ph | 1-methyl-1H-pyrazol-3-ylamino | 0.052 | 4.5 |
| 1h | 4-methoxy-3-pyridyl | cyclopropylamino | 0.25 | 16 (15)b |
| 3a | 6-acetamidoindol-1-yl | cyclopropylamino | 0.010 | 0.53 (1.0)b |
| 3b | 6-acetamidoindazol-1-yl | cyclopropylamino | 0.026 | 3.7 (5.0)b |
| 3c | 6-acetamido-pyrrolo[3,2-b]pyridin-1-yl | cyclopropylamino | <0.003 | 3.0 (2.1)b |
| 3d | 6-acetamidoindolin-1-yl | cyclopropylamino | 0.004 | 0.53 (0.73)b |
| 3e | 6-acetamido-(3,4-dihydro-2H-benzo[b][1,4]oxazin-4-yl)- | cyclopropylamino | 0.27 | ND |
| 3f | 6-acetamido-(1,2,3,4-tetrahydroquinolin-7-yl)- | cyclopropylamino | 0.24 | ND |
| 3g | 6-acetamido-5-methyl-indol-1-yl | cyclopropylamino | 0.079 | >30 |
| 3h | 6-acetamido-indol-1-yl | 1-methyl-1H-pyrazol-3-ylamino | 0.024 | 0.46 (0.56)b |
Mean value of three experiments. Deviations were within < ±25%.
SW480 Alamar Blue proliferation assay; ND = not determined.
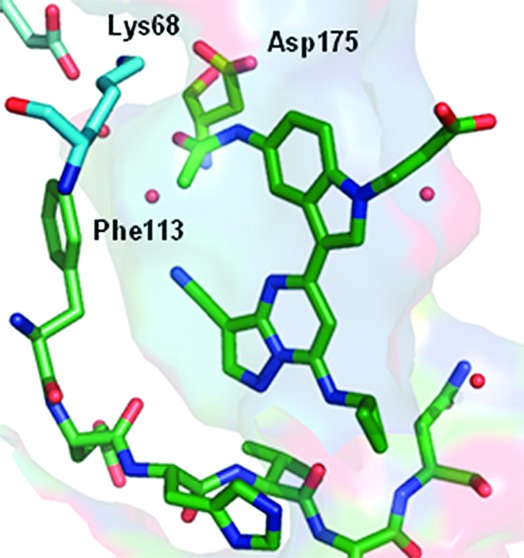
During the course of this effort, a disclosure16 by other researchers described a series of CK2 inhibitors (2) with pharmacophoric elements designed to establish interactions with Lys68, Asp175, and a previously observed structural water molecule.12 We sought to couple these insights with further modifications identified in our own work. In silico modeling of 1a with compound 2 (Figure 1) suggested the introduction of acetamide-containing 5,6-fused ring systems at the C5 position of the pyrazolo[1,5-a]pyrimidine core to give conformationally constrained hybrid structures such as 3a (Figure 2).
Figure 1.
Overlay of screening hit 1a with compound 2 illustrating their computationally derived binding poses in CK2.
Figure 2.

Design of fused-ring analogue 3a.
To test this design hypothesis, we prepared a series of 3-cyano-7-cyclopropylamino-pyrazolo[1,5-a]pyrimidines substituted at the 5-position with various 5,6- and 6,6-fused ring heterocycles (Table 1). Gratifyingly, indole 3a (CK2 IC50 = 10 nM) demonstrated that a planar, bicyclic structure is accommodated by the receptor and that the aniline NH of 2 is less important for its activity. Introduction of nitrogen into the five-membered ring, as with indazole 3b, led to a modest reduction in potency whereas 4-azaindole derivative 3c exhibited enhanced enzymatic potency relative to 3a. Partial saturation of the five-membered ring, as with indoline 3d, preserved activity; however, partially saturated 6,6-fused analogues such as benzoxazine 3e and tetrahydroquinoline 3f are 10-fold less active. Substitution adjacent to the acetamide, as in 5-methyl derivative 3g, is disfavored, presumably due to destabilization of the interaction with Lys68 and Asp175 via increased out-of-plane rotation. Deletion of the 3-cyano group in 3a (not shown) led to a significant reduction in enzymatic activity (CK2 IC50 = 2.6 μM).
Cellular potency for this series was determined using mechanistic and phenotypic end points. Depletion of cellular levels of pAKTS129, a direct substrate of CK2 believed to hyperactivate the AKT pathway,7 was measured in Western blot and ELISA (enzyme-linked immunosorbent assay) formats (Figure 3).17 Consistent with effects on prosurvival signaling, 3a induces apoptosis, as measured by caspase activation in HCT-116 cells (not shown). Indole 3a and indoline 3d possess comparable antiproliferative activity. However, for reasons not clear to us, indazole 3b and 4-azaindole derivative 3c exhibit a 30–100-fold drop in phenotypic potency relative to the effects observed in the pAKTS129 ELISA assay. Comparable potency was exhibited in a SW480 cell proliferation assay.
Figure 3.

(a) Concentration-dependent depletion of pAKTS129 in HCT-116 cells at 3 h and 24 h by 3a, as measured by Western blot analysis. (b) Inhibition of pAKTS129 in DLD-1 cells by 3h as determined by ELISA. (c) Depletion of pAKTS129 in DLD-1 cells upon treatment with 3h at 24 h as determined by Western blot.
The physical properties for 3a–h are poor, with low aqueous solubility, high protein binding in human plasma, and moderate lipophilicity (3a; solubility <10 μM at pH 7, fu <1%, log D = 3.3). As a consequence, the in vivo pharmacokinetic profile for 3a following a single oral 10 mg/kg dose in the rat is characterized by poor oral bioavailability (F = 5%) and low clearance (Cl = 10 mL/(min/kg)). In addition, 3a possesses moderate activity at the hERG (human Ether-a-go-go Related Gene) ion channel (hERG IC50 = 8.6 μM) and inhibits CYP1A2 (cytochrome p450 isozyme) with an IC50 = 0.1 μM. Conversely, indazole 3b and indoline 3d exhibit reduced hERG channel activity (hERG IC50 >33 μM) while 4-azaindole 3c is devoid of CYP1A2 activity (CYP1A2 IC50 > 20 μM).
We next attempted to improve the physical properties in 3a by replacing the N7 cyclopropyl substituent with hydrophilic groups. Analysis of the ATP-binding site suggested that several residues in the solvent channel are potentially disposed to form favorable interactions with polar groups and may also serve as selectivity determinants against other kinases. On the basis of the SAR for the indole and related heterocycles, we felt that we could optimize cellular potency and physical properties through modifications in other portions of the structure and potentially employ the indoline or 4-azaindazole rings to address in vitro ADME issues should they persist.
Replacement of the cyclopropyl group of 3a with four- and five-membered heterocycles is well-tolerated; N-methylpyrazole 3h exhibits cellular potency equivalent to that of 3a (pAKT IC50 = 16 nM) while oxetane 4a is 4-fold less active (Table 2). Hydrophilic substituents at the terminus of two- and three-atom spacers preserved enzymatic activity and enhanced physical properties but led to significant reduction in cellular potency. Despite possessing comparable enzymatic activity with 3h, 1-hydroxybutyl derivative 4b and dimethylamino-propyl derivative 4e exhibit a 10- and 100-fold drop in potency in the mode of the action assay (pAKTS129 ELISA), respectively. The permeability of 3a in MDCK-MDR1 (multidrug resistance protein 1) cells is moderate (A-B Papp = 22 × 10–6 cm/s) with low efflux characteristics (efflux ratio = 1.4). Conversely, both 4c and 4f exhibit low A-B permeability (A-B Papp ≤5 × 10–6 cm/s) and high efflux potential (efflux ratio = 17) in MDR1 overexpressing LLC/PK1 cells. In addition, compound 4f exhibits virtually no exposure (AUC = 0.065 μM·h) following administration of a single 10 mg/kg oral dose in the rat. These results suggest that membrane permeation in this series is impaired when lipophilicity is reduced such that log D < 2.9, and the effect may be compounded by the presence of a basic amine.
Table 2. CK2 Biochemical and Cellular Potency and Physical Property Data for Indoles 4a–4i.

| cpd | R7 | R6 | CK2 IC50 (μM)a | pAKT IC50 (μM)a | HCT-116 GI50 (μM)a | log D | sol (μM) | Hu PPB Fu (%) |
|---|---|---|---|---|---|---|---|---|
| 4a | oxetan-3-yl | AcNH | 0.004 | 0.057 | 2.2 (2.5)b | 2.2 | 1 | 12.4 |
| 4b | 4-hydroxybutyl | AcNH | 0.030 | 0.19 | 2.8 | 2.6 | 8 | 4.1 |
| 4c | 2-morpholinoethyl | AcNH | 0.03 | 0.46 | >30b | ND | 2 | 11 |
| 4d | 2-(pyrrolidin-1-yl)ethyl | AcNH | 0.04 | 0.78 | ND | 2.2 | 15 | 5.6 |
| 4e | 3-(dimethylamino)propyl | AcNH | 0.030 | 1.2 | 25b | 1.6 | 750 | 10 |
| 4f | 3-(pyrrolidin-1-yl)propyl | AcNH | 0.018 | 0.86 | 13b | 1.8 | >1000 | 6.7 |
| 4g | cyclopropyl | AcNMe | 0.011 | 0.034 | ND | 2.9 | <1 | 2.5 |
| 4h | cyclopropyl | HOCH2 | 0.12 | 0.83 | 10.1 | 3.1 | <1 | 1.9 |
| 4i | cyclopropyl | MeSO2CH2 | 0.007 | 0.083 | 16.4 | 2.5 | <1 | 1.8 |
Mean value of at least three experiments.
SW480 Alamar Blue cell proliferation assay; ND = not determined.
We next focused on modifying the acetamide to address the observed drop-off in cellular potency and the low bioavailability of nonbasic compounds. Substitution of the acetamide NH of 3a, to give N-methylacetamide 4g, preserved enzymatic and cellular activity but led to reduced stability when incubated with rat liver microsomes (Clint = 100 mL/(min/kg)). Installation of a hydroxymethyl group (4h) led to a significant reduction in enzymatic and cellular potency. Sulfone 4i exhibited high potency in the biochemical assay and moderate activity in the pAKT assay but lacked any significant antiproliferative effect. These results also confirm the pivotal role of the acetamide carbonyl of 3a and suggest that the NH is less critical and that other variations on the donor–acceptor group may be tolerated.
As an alternative to the solvent channel approach, we explored the use of a “reversed” indole scaffold to incorporate polar functionality (Table 3). The N1-methyl analogue (5a) and the unsubstituted 5b are potent CK2 inhibitors and exhibit submicromolar activity in the proliferation assay. More elaborate substitution at this position gave compounds (5c–f) with high enzyme activity but weaker effects in the mode of action (pAKTS129) and phenotypic assays.
Table 3. CK2 Biochemical and Cellular Potency for 5a–f.

| cpd | R1 | CK2 IC50 (nM)a | pAKT IC50 (μM)a | HCT-116 GI50 (μM)a |
|---|---|---|---|---|
| 5a | Me | 6 | 0.027 | 0.7 (1.1)b |
| 5b | H | 5 | 0.077 | 0.7 (0.7)b |
| 5c | 2-hydroxyethyl | <3 | 0.50 | 2.7 (4.5)b |
| 5d | 3-hydroxypropyl | <3 | 0.21 | 2.3 (3.2)b |
| 5e | CH2CH2CO2Me | 15 | ND | 6.6b |
| 5f | CH2CH2CO2H | <3 | 3.1 | ND |
Mean value of at least three experiments.
SW480 Alamar Blue cell proliferation assay; ND = not determined.
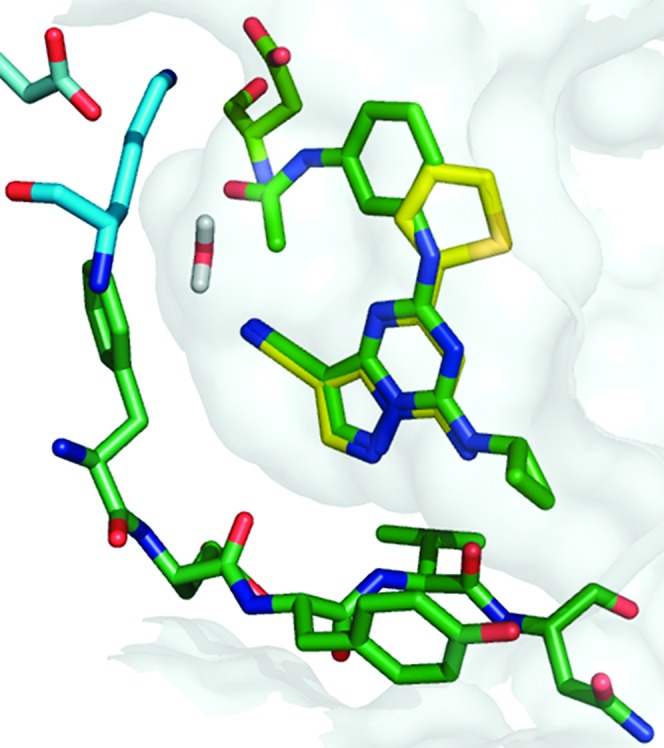
An X-ray cocrystal structure determination of 5f with recombinant human CK2 at 2.2 Å resolution showed the inhibitor bound as anticipated with N2 of the pyrazolo[1,5-a]pyrimidine core and the C7 NH interacting at the hinge region of the ATP binding pocket.17 As indicated in Figure 4, the energetic penalty associated with adopting the cis- conformation of the acetamide is compensated by a network of hydrogen bonding interactions with Lys68 and Asp175 via the carbonyl and NH, respectively. Moreover, a buried water molecule in the vicinity of the gatekeeper residue (Phe113) is coordinated by the acetamide carbonyl (2.7 Å) and the cyano group (3.0 Å). The propanoic acid adopts an extended conformation in which the β-CH2 group makes a hydrophobic contact between Gly46 and the side-chain of Val53 and positions the carboxylate group at the solvent-accessible surface.
Figure 4.

X-ray cocrystal structure determination of 5f with huCK2 at 2.2 Å. Bound water molecules are shown as spheres.
CK2 shares a high degree of binding site homology with a range of kinases, including GSK-3β (glycogen synthase kinase 3 beta) and CDK2 (cyclin-dependent kinase 2), targets for which pyrazolo[1,5-a]pyrimidine derivatives are reported to have affinity.18 Compound 3a was shown to be highly selective when tested against a panel of 324 kinases (Ambit Biosciences, San Diego, CA),19 at a concentration of 1 μM. From the 16 kinases with inhibition >50%, we selected four for subsequent IC50 determinations (Table 4). Introduction of nitrogen reduces affinity toward GSK-3β, as in indazole 3b (IC50 = 0.7 μM) and 4-azaindole 3c (IC50 = 1.0 μM). The presence of a basic group in the solvent channel region, as in 4e, reduces GSK-3β (IC50 = 2.6 μM) and Dyrk1 (dual-specificity tyrosine phosphorylation-regulated kinase 1) activity (IC50 = 2.6 μM). Members of the series exhibit a high degree of HipK2 (homeodomain-interacting protein kinase 2) potency, although substitution of the “reversed” indole NH reduces this affinity (5f, HipK2 IC50 = 0.57 μM). These data suggest that further enhancement of CK2 selectivity in this scaffold may be achieved through appropriate combination of the diversity elements identified thus far.
Table 4. Kinase Selectivity Data for 3a and Related Compounds.
| cpd | GSK-3β IC50 (μM) | HipK2 IC50 (μM) | Dyrk1 IC50 (μM) | CDK2 IC50 (μM) |
|---|---|---|---|---|
| 3a | 0.13 | 0.03 | 0.8 | 26 |
| 3b | 0.7 | 0.05 | 0.02 | >30 |
| 3c | 1.0 | 0.02 | 1.7 | >30 |
| 4e | 2.6 | 0.06 | 2.6 | >30 |
| 4g | 1.4 | <0.003 | 0.4 | >30 |
| 5b | 0.25 | <0.003 | 0.06 | 14 |
| 5f | 6 | 0.57 | 5.4 | >30 |
Synthesis of 5-acetamidoindole and related analogues (3a–h, 4a–i) was accomplished using a convergent approach (Scheme 1) that commenced with installation of the requisite amine by displacement of the 7-chloro group in 5,7-dichloro-3-cyanopyrazolo[1,5-a]pyrimidine (6). Replacement of the 5-chloro substituent in 7 was accomplished by Pd2(dba)3 (tris(dibenzylideneacetone)dipalladium(0))-mediated coupling via the NH of the appropriate fused ring system.20 In cases where the acetamide was not yet present in the coupling partner, the appropriate nitro precursor precursor (8, 11, 13) was utilized followed by subsequent reduction (to give 9, 12, and 14, respectively) and acetylation. Similarly, methylsulfonylmethyl derivative 4h was prepared from 4g followed by mesylation and displacement with MeSO2Na. Synthesis of “reversed” indoles (5a–f) was carried out beginning with the p-toluenesulfonyl derivative of 5-nitroindole (15) followed by conversion to mercury(II)acetate derivative 16 and subsequent treatment with borane to afford boronic acid 17.
Scheme 1.

Suzuki coupling of the latter with 7 afforded 18, which, following functional group manipulation, gave a mixture of 5a and 5b. Alkylation of the indole NH could be accomplished to deliver 5c and 5d. Conjugate addition of 5b with methyl methacrylate furnished 5e, which afforded 5f following ester hydrolysis (Scheme 2).
Scheme 2.

In conclusion, we have identified a series of potent and selective inhibitors of CK2 kinase using structure-based hybridization of an early screening hit with a literature scaffold. Enhancement of physical properties in the 6-acetamidoindole series (3a) was achieved by modification of the N7 substituent but had a deleterious effect on cellular potency. Kinase selectivity profiling has revealed 3a to be highly selective and has identified structural features that further reduce off-target effects. Exploitation of the crystallographically determined binding mode of 5f for enhancement of primary target potency and kinase selectivity will be described in a subsequent communication.
Supporting Information Available
Representative experimental procedures for synthesis of analogues, biochemical and cellular assays, and X-ray structure determination. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Pinna L. A. Protein kinase CK2: a challenge to canons. J. Cell Sci. 2002, 115, 3873–3878. [DOI] [PubMed] [Google Scholar]
- Allende J. E.; Allende C. C. Protein kinases 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J. 1995, 9, 313–323. [DOI] [PubMed] [Google Scholar]
- Ji H.; Wang J.; Nika H.; Hawke D.; Keezer S.; Ge Q.; Fang B.; Fang X.; Litchfield D. W.; Aldape K. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-catenin from beta-catenin and transactivation of beta-catenin. Mol. Cell 2009, 36, 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou D. Y.; Dominguez I.; Toselli P.; Landesman-Bollag E.; O’Brien C.; Seldin D. C. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol. Cell. Biol. 2008, 28, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchou T.; Vernet M.; Blond O.; Jensen H. H.; Pointu H.; et al. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol. Cell. Biol. 2003, 23, 908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzene M.; Pinna L. A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells?. Biochim. Biophys. Acta 2010, 1804, 499–504. [DOI] [PubMed] [Google Scholar]
- Di Maira G.; Brustolon F.; Pinna L. A.; Ruzzene M. Dephosphorylation and inactivation of Akt/PKB is counteracted by protein kinase CK2 in HEK 293T cells. Cell. Mol. Life Sci. 2009, 66, 3363–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez I.; Sonenshein G. E.; Seldin D. C. CK2 and its role in Wnt and NF-kappa B signaling: Linking development and cancer. Cell. Mol. Life Sci. 2009, 66, 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landesman-Bollag E.; Romieu-Mourez R.; Song D. H.; Sonenshein G. E.; Cardiff R. D.; Seldin D. C. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 2001, 20, 3247–3257. [DOI] [PubMed] [Google Scholar]
- Slaton J. W.; Unger G. M.; Sloper D. T.; Davis A. T.; Ahmed K. Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol. Cancer Res. 2004, 2, 712–721. [PubMed] [Google Scholar]
- Duncan J. S.; Litchfield D. W. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 2007, 1784, 33–47. [DOI] [PubMed] [Google Scholar]
- Battistutta R. Structural basis of protein kinase CK2 inhibition. Cell. Mol. Life Sci. 2009, 66, 1868–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre F.; Chua P. C.; O’Brien S. E.; Siddiqui-Jain A.; Bourbon P.; Haddach M.; Michaux J.; Nagasawa J.; Schwaebe M. K.; Stefan E.; Vialettes A.; Whitten J. P.; Chen T. K.; Darjania L.; Stansfield R.; Anderes K.; Bliesath J.; Drygin D.; Ho C.; Omori M.; Proffitt C.; Streiner N.; Trent K.; Rice W. G.; Ryckman D. M. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–54. [DOI] [PubMed] [Google Scholar]
- See Supporting Information.
- Bembenek S. D.; Tounge B. A.; Reynolds C. H. Ligand efficiency and fragment-based drug discovery. Drug Discovery Today 2009, 14, 278–283. [DOI] [PubMed] [Google Scholar]; LE (ligand efficiency) = −log(CK2 IC50)/number of heavy atoms.
- Nie Z.; Perretta C.; Erickson P.; Margosiak S.; Almassy R.; Lu J.; Averill A.; Yager K. M.; Chu S. Structure-based design, synthesis, and study of pyrazolo[1,5-a][1,3,5]triazine derivatives as potent inhibitors of protein kinase CK2. Bioorg. Med. Chem. Lett. 2007, 17, 4191–4195. [DOI] [PubMed] [Google Scholar]
- The PDB deposition code for the cocrystal structure is 3U4U.
- Williamson D. S.; Parratt M. J.; Bower J. F.; Moore J. D.; Richardson C. M.; Dokurno P.; Cansfield A. D.; Francis G. L.; Hebdon R. J.; Howes R.; Jackson P. S.; Lockie A. M.; Murray J. B.; Nunns C. L.; Powles J.; Robertson A.; Surgenor A. E.; Torrance C. J. Structure-guided design of pyrazolo[1,5-a]pyrimidines as inhibitors of human cyclin-dependent kinase 2. Bioorg. Med. Chem. Lett. 2005, 15, 863–867. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Kranenburg M.; van der Burgt Y. E. M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Goubitz K.; Fraanje J. New diphosphine ligands based on heterocyclic aromatics inducing very high regioselectivity in rhodium-catalyzed hydroformylation: effect of the bite angle. Organometallics 1995, 14, 3081–3089. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.