

Abstract

A series of (+)-negamycin 1 analogues were synthesized, and their readthrough-promoting activity was evaluated for nonsense mutations in Duchenne muscular dystrophy (DMD). A structure–activity relationship study indicated that 11b was the most potent drug candidate. Immunohistochemical analyses suggested that treatment with 11b restored dystrophin expression in mdx mice, a DMD mouse model. Furthermore, 11b decreased serum creatine kinase (CK) levels, an indicator of muscle fiber destruction. Most importantly, 11b demonstrated lower toxicity than 1, and thus, it could be a useful candidate for long-term treatment of DMD.
Keywords: Negamycin, readthrough-promoting activity, Duchenne muscular dystrophy, nonsense mutations, hydrazino dipeptide, genetic disease
Duchenne muscular dystrophy (DMD), characterized by progressive muscle degeneration, is one of the most common hereditary disorders, affecting approximately 1 in 3500 live male births.1 This disorder is caused by mutations in the DMD gene, located on the X-chromosome. The DMD gene encodes the protein dystrophin, which plays a crucial role linking the intracellular cytoskeleton and the extracellular matrix via the dystrophin-associated protein complex (DAPC). The loss of dystrophin function causes destabilization of the DAPC, which results in the breakdown of muscle fibers, loss of membrane integrity, and difficulty in walking and breathing, and it ultimately leads to death. Nonsense mutations, which lead to premature termination codons (PTCs) in the reading frame of the DMD gene, are responsible for up to 20% of DMD cases. The nonsense mutations yield truncated dystrophin proteins, which have no valuable biological function.2 Presently, although the molecular basis for the disease is clear, there is no cure for DMD.3 The only available treatment is glucocorticoid therapy, which can prolong ambulation and reduce the incidence of severe scoliosis, although it is limited to relatively short-term treatments due to severe side effects.4−6
Recently, a unique therapeutic strategy, so-called “readthrough drugs”, was proposed to target genetic diseases caused by nonsense mutations.7 These drugs promote a translational “skip” of PTCs, but not of normal termination codons, resulting in the production of full-length proteins. Specifically, gentamicin, an aminoglycoside antibiotic, was reported to promote the readthrough of disease-causing PTCs in mammalian cells. Furthermore, its treatment partially restored dystrophin expression in skeletal and cardiac muscles of mdx mice, an animal model of DMD with a nonsense mutation in the dystrophin gene. In spite of these positive results, long-term administration of gentamicin is not recommended, due to its severe side-effects including ototoxicity8 and nephrotoxicity.9 Small molecules possessing readthrough-promoting activity have also been described for DMD treatment, including aminoglycosides,10 RTC compounds,11 and an oxadiazole derivative, ataluren (PTC-124, phase IIB).12
In the same vein, Arakawa et al.13 reported that the dipeptidic antibiotic (+)-negamycin (1, [2-(3,6-diamino-5-hydroxyhexanoyl)-1-methylhydrazino]acetic acid, Figure 1)14 also induced the readthrough of PTCs in both a prokaryotic translational system15 and mdx mice.13 Therefore, 1 has been recognized as a potential therapeutic agent for diseases caused by nonsense mutations. Here, we designed and synthesized a series of negamycin analogues, and their biological activity was evaluated using a transgenic mouse strain, READ (readthrough evaluation and assessment by dual reporter),16 which expresses a dual-reporter gene segmentalized with a PTC. Once the most potent 11b had been identified (Table 1), we used mdx mice to assess the effect on dystrophin expression, serum creatine kinase levels,17 which are a clinical indicator of DMD, and general toxicity. We found that 11b performed better than 1, with markedly reduced toxicity, thus making it a promising therapeutic candidate.
Figure 1.
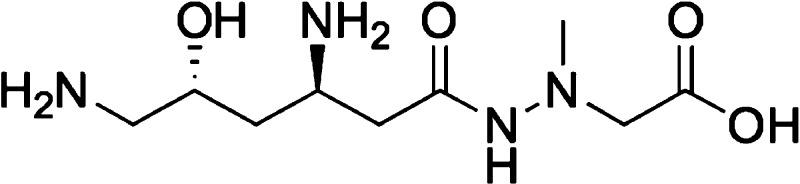
Structure of (+)-negamycin 1.
Table 1. Readthrough-Promoting and Antimicrobial Activities of Synthetic Negamycin Analogues.

Synthetic yields were calculated from intermediates 3 or 7 for analogues 6 or 11a–f and 14, respectively.
NA; not applicable, see ref (18).
Relative in vivo readthrough-promoting activity, which is expressed as a ratio compared to gentamicin. Samples were subcutaneously injected at the abdominal region of the READ mouse with a dosage of 0.1 mg/day/20 g body-weight for 7 days. Data are mean ± SD (n = 4).
The antimicrobial activity (MIC) against several microorganisms (Staphylococcus aureus FDA 209P/Bacilus subtilis NRRL B-558/Escherichia coli BEM11/Shigella dysenteriae J S11910/Pseudomonas aeruginosa A3, respectively). “– ” denotes >128 μg/mL (MIC). See ref (24).
NT: not tested.
(+)-Negamycin 1 was first isolated in 1970 from a microorganism closely related to Streptomyces purpeofuscus.14 In an attempt to synthesize chiral 1, we developed shortened, highly efficient synthetic routes.18,19 Using one of these routes as a starting point,19 here, we synthesized a series of analogues. Briefly, for the synthesis of analogue 6 (Scheme 1A), intermediate 3 was prepared from the commercially available ester 2 over 7 steps.19 Then, 3 was converted to the N-protected tert-butyl ester 4 as a single diastereomer (diastereomeric excess (de) >99%) over 3 steps using Node’s asymmetric Michael addition,20 removal of the chiral auxiliary,21 and protection of the inserted 3-amino group. The obtained intermediate 4 was then efficiently converted to the acid form by a microwave-assisted saponification, and it was subsequently coupled with a hydrazino ester using an EDC-HOBt (EDC, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; HOBt, 1-hydroxybenzotriazole) method22 to yield 5. Deprotection of 5 with 4 M HCl/dioxane and purification by ion exchange chromatography afforded 6 (specific rotation: found [α]D26.4 +14.0 (c 0.94, H2O), literature data23 [α]D22.0 +8.5 (c 0.70, H2O)).
Scheme 1. Synthesis of Analogue 6.

Reagents and conditions: (A) Synthesis of 6: (a) (i) KOH, MeOH, microwave (300 W), 100 °C, 10 min; (ii) PTSA·H2N–N(Me)CH2CO2t-Bu, HOBt·H2O, Et3N, EDC·HCl, CH2Cl2, rt, 4 h, 62% (2 steps); (b) (i) 4 M HCl/dioxane, rt, 1 h; (ii) ion exchange chromatography, 98%. (B) Synthesis of 11a–f: (a) (i) DIBAL-H, toluene, −78 °C, 2 h; (ii) Ph3P = CHCO2Bn, THF, reflux, overnight, 69% (2 steps); (b) Pd/C, H2, MeOH, rt, 1.5 h, quant; (c) amino acid t-Bu esters or hydrazinoacid t-Bu ester, HOBt·H2O, Et3N, EDC·HCl, DMF, rt, 3 h to overnight, 58–95%; (d) (i) 4 M HCl/dioxane, rt, 1 h; (ii) reversed-phase HPLC, 30–96%. (C) Synthesis of 14: (a) (i) DIBAL-H, toluene, −78 °C, 2 h; (ii) Ph3P = CHCO2Me, THF, reflux, overnight, 63% (2 steps); (b) (i) KOH, MeOH/H2O (2:1), rt, 4 h; (ii) HCl·H-Gly-Ot-Bu, HOBt·H2O, Et3N, EDC·HCl, DMF, rt, overnight, 44% (2 steps); (c) (i) 4 M HCl/dioxane, rt, 1 h; (ii) reversed-phase HPLC, 85%.
Next, analogues 11a–f were synthesized (Scheme 1B). Weinreb amide 7(19) was prepared from 2 over six steps, and then it was reduced with diisobutylaluminium hydride (DIBAL-H) to the corresponding aldehyde, directly followed by treatment with (benzyloxycarbonylmethylene)triphenylphosphorane in THF under reflux conditions. After purification by flash chromatography on silica (Silica Gel 60N, KANTO CHEMICAL), we obtained 8 in 69% yield over two steps. After 8 was treated with Pd/C under a H2 atmosphere, the resultant 9 was coupled with various amino acid tert-butyl esters or a hydrazinoacid tert-butyl ester18 using an EDC-HOBt method to obtain 10a–f. Deprotection of 10a–f with 4 M HCl/dioxane and purification by reversed-phase HPLC afforded 11a–f with 30–96% yield.
In the synthesis of analogue 14 (Scheme 1C), 7 was converted to the intermediate 12 by a procedure similar to that employed for 8 (Scheme 1B). Then, 12 was converted to the acid form by saponification and subsequently coupled with HCl·H-Gly-Ot-Bu using an EDC-HOBt method to yield 13. Deprotection of 13 with 4 M HCl/dioxane and purification by reversed-phase HPLC afforded 14 with 85% yield. The purity of each synthesized analogue for biological evaluation was over 95%.
To evaluate the readthrough-promoting activity, we adapted an in vivo dual-reporter gene expression system using READ mice.16 This system encodes β-galactosidase and luciferase genes connected with a PTC (see the Supporting Information). β-Galactosidase activity is present constitutively, but luciferase activity is only detected when readthrough occurs. Therefore, the activities of both enzymes in skeletal muscle were measured to calculate the activity ratio of luciferase to β-galactosidase after negamycin analogues (0.1 mg) were subcutaneously administered in the abdominal region of READ mice for 7 days. The antimicrobial activity was also measured.24 The results of these biological evaluations are shown in Table 1.
Since synthetic 1 showed similar levels of readthrough-promoting activity to the extracted native 1 (data not shown) and gentamicin, we first evaluated the importance of stereochemistry at the 3-amino group. The (+)-3-epi-negamycin 6 exhibited equipotent activity to 1, suggesting that the stereochemistry of the 3-amino group might not be important for the activity. Next, analogue 11a with no 3-amino group was prepared (Table 1). However, complete removal of the amino group led to a decrease and a loss of the readthrough-promoting and antimicrobial activities, respectively. Thus, the presence, but not the stereochemistry, of the 3-amino group was important for both biological activities.
In striking contrast, however, we observed that when both the N-methyl and amino groups were omitted from 11a, the corresponding glycine analogue 11b was a potent promoter of readthrough activity, demonstrating a 1.4-fold increase in functionality as compared to the case of 1. Importantly, 11b also did not display antimicrobial activity, making it a more selective readthrough-promoting analogue than 1. In other words, it means that the readthrough-promoting activity can be distinguished from the antimicrobial activity.
Encouraged by these results, we synthesized additional analogues based on the chemical structure of 11b. However, both the ethyl ester analogue 11c and the N-methyl glycine analogue 11d demonstrated decreased activities. From these results, we inferred that the glycine residue with a free carboxylic acid was functionally important, a hypothesis that we confirmed using 11e and 11f. Moreover, 14, with the unsaturated amide structure, did not show any significant activity.
To understand the biological effects in detail, the most active 11b was chosen for further in vivo immunohistochemical and biochemical evaluations. Regarding the immunohistochemical evaluation, 11b was subcutaneously injected in the abdominal region of mdx mice at a dosage of 1 mg in phosphate-buffered saline (PBS, 0.2 mL)/day/20 g body-weight for 4 weeks. Dystrophin expression was clearly observed in the skeletal muscle of wild-type B10 mice (Figure 2A), while mdx mice lacked this signal (Figure 2B). In contrast to these controls, dystrophin expression was only partially restored in the skeletal muscle of 11b-treated mdx mice (Figure 2C). However, this result suggested that 11b promotes PTC readthrough and is therefore a potential therapeutic candidate for DMD.
Figure 2.
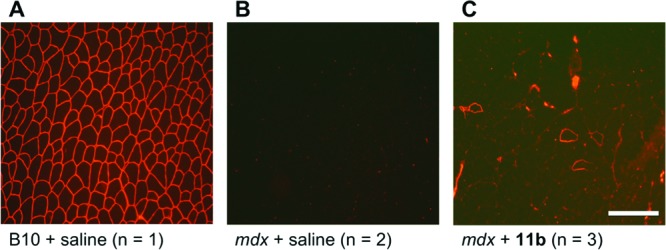
Dystrophin expression in skeletal muscles. Immunofluorescent staining of dystrophin in mouse muscle tissues was performed on 8 μm transverse cryosections.12 (A) wild-type B10 mouse; (B) untreated mdx mouse; (C) 11b-treated mdx mouse. Bar = 200 μm.
Regarding biochemical evaluation, we assessed levels of serum creatine kinase (CK)17 in mdx mice treated subcutaneously with 11b at a dosage of 1 mg in PBS (0.2 mL)/day/20 g body-weight for 4 weeks. As controls, the CK level in a wild-type B10 mouse was very low, while levels in mdx mice were very high. A statistically significant reduction of serum CK levels in 11b-treated mdx mice was observed in comparison to the case of the untreated controls (Figure 3A). This result suggested that 11b could enhance the strength of muscle fibers by increasing functional protein expression.
Figure 3.

(A) Serum CK levels in mdx mice: wild-type B10 (n = 1); mdx (n = 2); 11b-treated mdx (n = 3). (B) Effect of 11b on the body-weight of mdx mice. The body-weight of 1- and 11b-treated mice (n = 4, 1 mg/day/20 g body-weight) over the course of 4 weeks was measured in comparison to that of saline-treated mice (n = 2) as a control. (C) Effects of the administration of high doses of 11b on the readthrough-promoting activity in READ mice. P: Probability-value. Error bar indicates SD.
Next, we examined the acute in vivo toxicity of 11b as compared to 1 by measuring the body-weight change of mdx mice for 4 weeks. Improving the in vivo toxicity profile of 1 was an important goal for the development of readthrough drugs based on the negamycin structure. As shown in Figure 3B, over the course of 4 weeks, the body-weight of saline-treated mice gradually increased, while that of 1-treated mice (1 mg in 0.2 mL saline/day/20 g body-weight) markedly decreased. Conversely, the body-weight of 11b-treated mice (1 mg in 0.2 mL saline/day/20 g body-weight) slowly increased during this time frame, indicating that 11b exhibited a lower toxicity profile than 1. We postulate that this lower toxicity is due to the absence of the hydrazine structure in 11b. This improved toxicity profile strongly supports the potential of 11b for the long-term treatment of DMD.
Finally, inspired by the low toxicity observed with 11b, we tested the effects of high doses of 11b on PTC readthrough-promoting activity. Accordingly, 11b, or gentamicin or saline, as positive and negative controls, respectively, was administered subcutaneously in READ mice for 7 days. As shown in Figure 3C, the readthrough-promoting activity of 11b was not dose-dependent at the levels tested. However, at a 3 mg dose, 11b was more effective than gentamicin. For unknown reasons, there appeared to be a reduction in readthrough-promoting activity at the highest dosage of 11b, an observation that we will pursue in the near future.
In summary, we have synthesized a series of (+)-negamycin analogues and evaluated their readthrough-promoting activity for DMD. On the basis of SAR studies, we identified 11b as the most potent candidate. This analogue was then taken forward through immunohistochemical and biochemical studies, which demonstrated that treatment with 11b restored some dystrophin expression in mdx mice and decreased their serum CK levels, indicating that the drug was protecting muscular tissues from collapse. Most importantly, 11b was shown to have a lower toxicity profile than 1, which might be useful for the long-term treatment of DMD. Further SAR studies to develop more efficient derivatives are under investigation.
Acknowledgments
The authors thank Ms. Miyuki Hiroshima and Ms. Yumi Sakurai, Tokyo University of Pharmacy and Life Sciences, for technical assistance and Dr. Minoru Ozeki and Emeritus Professor Manabu Node, Kyoto Pharmaceutical University, for technical advice regarding the asymmetric Michael addition.
Supporting Information Available
Synthetic procedures, characterization of new products, biological assay protocols, and NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Sources
This research was supported by an Intramural Research Grant (23-5) for Neurological and Psychiatric Disorders of NCNP, a Grant-in-Aid for Scientific Research (B) (No. 20390036) from MEXT (Ministry of Education, Culture, Sports, Science and Technology), the Ichiro Kanehara Foundation, Sasakawa Grants for Science Follows (to M.S.) partly, and a Health and Labor Sciences Research Grant for Research, Comprehensive Research on Disability Health and Welfare (19A-020 and H22-016) from the Ministry of Health, Labor and Welfare, Japan.
The authors declare no competing financial interest.
Supplementary Material
References
- Nowak K. J.; Davies K. E. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep. 2004, 5, 872–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik V.; Rodino-Klapac L. R.; Viollet L.; Mendell J. R. Aminoglycoside-induced mutation suppression as a therapeutic strategy for Duchenne muscular dystrophy. Ther. Adv. Neurol. Disord. 2010, 3, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco A. P.; Neve R. L.; Colletti-Feener C.; Bertelson C. J.; Kurnit D. M.; Kunkel L. M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [DOI] [PubMed] [Google Scholar]
- Granchelli J. A.; Pollina C.; Hudecki M. S. Pre-clinical screening of drugs using the mdx mouse. Neuromuscular Disord. 2000, 10, 235–239. [DOI] [PubMed] [Google Scholar]
- Griggs R. C.; Moxley R. T. III; Mendell J. R.; Fenichel G. M.; Brooke M. H.; Pestronk A.; Miller J. P.; Cwik V. A.; Pandya S.; Robinson J. Duchenne dystrophy: randomized, controlled trial of prednisone and azathioprine. Neurology 1993, 43, 520–527. [DOI] [PubMed] [Google Scholar]
- Khurana T. S.; Davies K. E. Pharmacological strategies for muscular dystrophy. Nat. Rev. Drug Discovery 2003, 2, 379–390. [DOI] [PubMed] [Google Scholar]
- Barton-Davis E. R.; Cordier L.; Shoutourma D. I.; Leland S. E.; Sweeney H. L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Invest. 1999, 104, 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchin T.; Cortopassi G. Proposed molecular and cellular mechanism for aminoglycoside ototoxicity. Antimicrob. Agents Chemother. 1994, 38, 2517–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingeot-Leclercq M. P.; Tulkens P. M. Aminoglycosides: Nephrotoxicity. Antimicrob. Agents Chemother. 1999, 43, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudleman I.; Rebibo-Sabbah A.; Cherniavsky M.; Belakhov V.; Hainrichson M.; Chen F.; Schacht J.; Pilch D. S.; Ben-Yosef T.; Baasov T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 2009, 52, 2836–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.; Damoiseaux R.; Nahas S.; Gao K.; Hu H.; Pollard J. M.; Goldstine J.; Jung M. E.; Henning S. M.; Bertoni C.; Gatti R. A. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J. Exp. Med. 2009, 206, 2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch E. M.; Barton E. R.; Zhuo J.; Tomizawa Y.; Friesen W. J.; Trifillis P.; Paushkin S.; Patel M.; Trotta C. R.; Hwang S.; Wilde R. G.; Karp G.; Takasugi J.; Chen G.; Jones S.; Ren H.; Moon Y.; Corson D.; Turpoff A. A.; Campbell J. A.; Conn M. M.; Khan A.; Almstead N. G.; Hedrick J.; Mollin A.; Risher N.; Weetall M.; Yeh S.; Branstrom A. A.; Colacino J. M.; Babiak J.; Ju W. D.; Hirawat S.; Northcutt V. J.; Miller L. L.; Spatrick P.; He F.; Kawana M.; Feng H.; Jacobson A.; Peltz S. W.; Sweeney H. L. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [DOI] [PubMed] [Google Scholar]
- Arakawa M.; Shiozuka M.; Nakayama Y.; Hara T.; Hamada M.; Kondo S.; Ikeda D.; Takahashi Y.; Nonomura Y.; Sheykholeslami K.; Kondo K.; Kaga K.; Kitamura T.; Suzuki-Miyagoe Y.; Takeda S.; Matsuda R. Negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice. J. Biochem. 2003, 134, 751–758. [DOI] [PubMed] [Google Scholar]
- Hamada M.; Takeuchi T.; Kondo S.; Ikeda Y.; Naganawa H.; Maeda K.; Okami Y.; Umezawa H. A new antibiotic, negamycin. J. Antibiot. 1970, 23, 170–171. [DOI] [PubMed] [Google Scholar]
- Uehara Y.; Hori M.; Umezawa H. Negamycin inhibits termination of protein synthesis directed by phage f2 RNA in vitro. Biochim. Biophys. Acta 1974, 374, 82–95. [DOI] [PubMed] [Google Scholar]
- Shiozuka M.; Wagatsuma A.; Kawamoto T.; Sasaki H.; Shimada K.; Takahashi Y.; Nonomura Y.; Matsuda R. Transdermal delivery of a readthrough-inducing drug: a new approach of gentamicin administration for the treatment of nonsense mutation-mediated disorders. J. Biochem. 2010, 147, 463–470. [DOI] [PubMed] [Google Scholar]
- Ebashi S.; Toyokuma Y.; Momoi H.; Sugita H. High creatine phosphokinase activity of sera of progressive muscular dystrophy. J. Biochem. 1959, 46, 103–104. [Google Scholar]
- Hayashi Y.; Regnier T.; Nishiguchi S.; Magne O. S.; Hashimoto D.; Hasegawa J.; Katoh T.; Kajimoto T.; Shiozuka M.; Matsuda R.; Node M.; Kiso Y. Efficient total synthesis of (+)-negamycin, a potential chemotherapeutic agent for genetic diseases. Chem. Commun. 2008, 2379–2381. [DOI] [PubMed] [Google Scholar]
- Nishiguchi S.; Magne O. S.; Taguchi A.; Regnier T.; Kajimoto T.; Node M.; Yamazaki Y.; Yakushiji F.; Kiso Y.; Hayashi Y. Total synthesis of (+)-negamycin and its 5-epi-derivative. Tetrahedron 2010, 66, 314–320. [Google Scholar]
- Node M.; Hashimoto D.; Katoh T.; Ochi S.; Ozeki M.; Watanabe T.; Kajimoto T. Asymmetric Michael addition of a recyclable chiral amine: inversion of stereoselectivity caused by the difference of ethereal solvents. Org. Lett. 2008, 10, 2653–2656. [DOI] [PubMed] [Google Scholar]
- Katoh T.; Watanabe T.; Nishitani M.; Ozeki M.; Kajimoto T.; Node M. Selective C-N bond oxidation: demethylation of N-methyl groups in N-arylmethyl-N- methyl-α-amino esters utilizing N-iodosuccinimide (NIS). Tetrahedron Lett. 2008, 49, 598–600. [Google Scholar]
- Konig W.; Geiger R. A. A new method for synthesis of peptides: activation of the carboxyl group with dicyclohexylcarbodiimide using 1-hydroxybenzotriazoles as additives. Chem. Ber. 1970, 103, 788–798. [DOI] [PubMed] [Google Scholar]
- Davies S. G.; Ichihara O.; Robert P. M.; Thomson J. E. Asymmetric syntheses of (+)-negamycin, (+)-3-epi-negamycin and sperabillin C via lithium amide conjugate addition. Tetrahedron 2011, 67, 216–227. [Google Scholar]
- The antimicrobial activity (MIC) was measured with the routine screening system at the Institute of Microbial Chemistry (IMC), Tokyo, Japan, using the agar dilution streak method (2-fold dilution) in Mueller Hinton agar (Difco) at 37 °C for 18 h against 34 microorganisms.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.