

Abstract

We have discovered a novel series of 4-azetidinyl-1-aryl-cyclohexanes as CCR2 antagonists. Divergent SAR studies on hCCR2 and hERG activities led to the discovery of compound 8d, which displayed good hCCR2 binding affinity (IC50, 37 nM) and potent functional antagonism (chemotaxis IC50, 30 nM). It presented an IC50 of >50 μM in inhibition of the hERG channel and had no effect on the QTc interval up to 10 mg/kg (i.v.) in anesthetized guinea pig and dog CV studies. It also displayed high selectivity over other chemokine receptors and GPCRs, and amendable oral bioavailability in dogs and primates. In a thioglycollate-induced inflammation model in hCCR2KI mice, it had ED50 of 3 mg/kg on inhibition of the influx of leukocytes, monocytes/macrophages, and T-lymphocytes.
Keywords: Azetidine, CCR2, hERG, cardiovascular (CV) safety
Monocyte chemotactic protein-1 (MCP-1) is one of the primary molecules controlling the influx of mononuclear leukocytes into sites of inflammation.1,2 MCP-1 is a potent chemotactic factor for monocytes and memory T lymphocytes, and it stimulates the movement of those cells along a chemotactic gradient following binding to its cell-surface receptor, CC chemokine receptor-2 (CCR2). This ligand/receptor pair is overexpressed under numerous inflammatory conditions wherein excessive monocyte recruitment is observed. Indeed, CCR2- and MCP-1-deficient mice and CCR2 or MCP-1 antibody-treated rodents show decreased recruitment of monocytes and produce markedly attenuated inflammatory responses in animal models of rheumatoid arthritis (RAs),3 multiple sclerosis,4 asthma,5 atherosclerosis,6 diabetes,7 allograft rejection,8 and neuropathic pain.9 Clearly, these observations confirm the role of CCR2 in the pathogenesis of several immune-based inflammatory diseases and identify this chemokine receptor as a potentially valuable therapeutic target. Thus, an antagonist of the binding of MCP-1 to its receptor may be an effective treatment for any inflammatory disease in which monocytes, mast cells, or basophils play major roles.
While a leading CCR2 antagonist MK-0812 failed to show significant improvement on RAs in clinical trials, there has been continuously intensive research interest in many distinct chemical series as CCR2 antagonists for other indica-tions.10−17 We have recently reported a novel series of 4-azetidinyl-1-arylcyclohexanes as CCR2 antagonists.16 This scaffold is characterized by a general hERG liable structure consisting of a central basic amine flanked by two hydrophobes at the ends. Divergent SARs on hCCR2 and hERG activities enabled us to dial out hERG affinity and generated highly selective hCCR2 antagonists in the scaffold. The lead compound 1 (Figure 1) possessed good hCCR2 binding affinity (IC50, 15 nM), potent functional activity (chemotaxis, IC50, 22 nM), and amendable separation over hERG activities. Disappointingly, i.v. treatment of anesthetized guinea pigs with 1 resulted in a significant dose dependent prolongation of the QTc interval (ΔQTc). At a dose of 10 mg/kg, a +20% ΔQTc was observed, with a drug plasma level of 12 μM being attained. Thus, 1 was abandoned from further development due to its unacceptable CV safety profile. Since QTc prolongation has been linked to preferential blockade of the voltage-gated potassium (K+) channel encoded by the hERG, optimization of a structural series for lack of in vitro hERG affinity appears to be generally predictive of decreased potential to cause QTc prolongation in vivo. Herein, we report our continuous SAR studies on identifying potent and selective hCCR2 antagonists devoid of this ancillary cardiac activity. Owing to the correlation between the ΔQTc observed with 1 in guinea pigs and the potency of 1 in the hERG binding assay, we defined hERG IC50’s > 25 μM must be achieved to address the QTc issue. As a critical follow-up, a hERG patch clamp assay was used as a second front-line screening for eliminating compounds that displayed significant blockade of the potassium current, which is another in vitro indicator for causing QTc prolongation.
Figure 1.
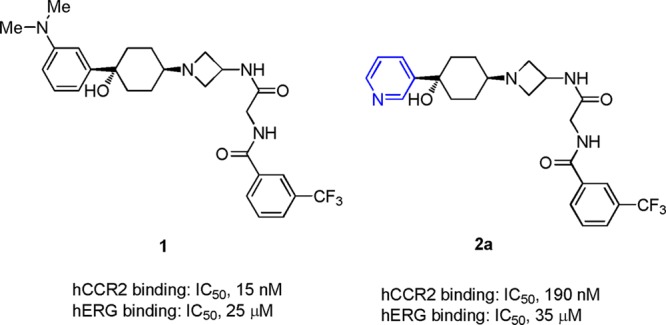
Early leads.
In the discovery of lead compound 1, it was established that the cyclohexyl azetidine core as well as the right-end bis-amide functionality were critical for hCCR2 activity and might be too sensitive for modifications.16 The left-end phenyl ring was somehow tolerated for moderate to good hCCR2 activity. For this reason, we focused further optimization on the left-end aromatic group. Replacement of the phenyl group at the 1-position of the cyclohexyl ring with the 3-pyridinyl group resulted in a significant loss of hCCR2 activity but also further attenuated hERG binding affinity (2a: hCCR2 IC50, 190 nM; hERG IC50, 35 μM).15−17 These data prompted us to take nitrogen containing six membered aromatic rings as the starting point for subsequent optimization (Table 1). Compound 2b, bearing a more polar pyrimidinyl group, displayed even weaker hCCR2 binding affinity (IC50, 650 nM) compared with 2a. We then examined the substituent effect on both hCCR2 and hERG inhibition among the pyridinyl derivatives. Electron withdrawing groups such as fluoro or cyano did not improve hCCR2 binding, as indicated with 2c and 2d, with IC50 values of 190 nM and 170 nM, respectively. In contrast, the electron donating group, exemplified by methoxy and methyl groups, had a beneficial effect on hCCR2 activity, as evidenced by an approximate 3–5-fold increase of binding affinity (2e and 2f vs 2a). The improved potency of 2e and 2f was also translated into good levels of activity in the chemotaxis assay with IC50 values of 74 nM and 56 nM, respectively. To our delight, installation of both substituents maintained good selectivity between hCCR2 and hERG binding affinities (hERG binding IC50 of 35 and 37 μM). The weak hERG affinities of both compounds were further confirmed with only 12% and 29% of inhibition at 3 μM in the patch clamp assay. The dimethylamino analogue 2g was ∼8-fold weaker for hCCR2 binding than 2f. Addition of a methyl group ortho to the methoxy group also resulted in significant loss of hCCR2 affinity, as shown by 2h (IC50, 220 nM). The two methoxy-pyridinyl regioisomers 2i and 2j displayed weaker hCCR2 binding affinities (IC50, 71 and 250 nM) compared with that of 2f, indicating that the electronic density of the pyridinyl ring had a great impact on the hCCR2 activity. As expected, installation of more lipophilic 4-alkoxy groups maintained or improved both hCCR2 binding affinity and chemotaxis activity, as illustrated by 2k–2o. Steric bulkiness on the alkoxy group was well tolerated for good hCCR2 activity (2m, 2n, 2o). Unfortunately, the unwanted hERG signals among these derivatives reappeared to be problematic for further evaluations.
Table 1. SAR of Nitrogen Containing Six Membered Heterocyclic Analogues.
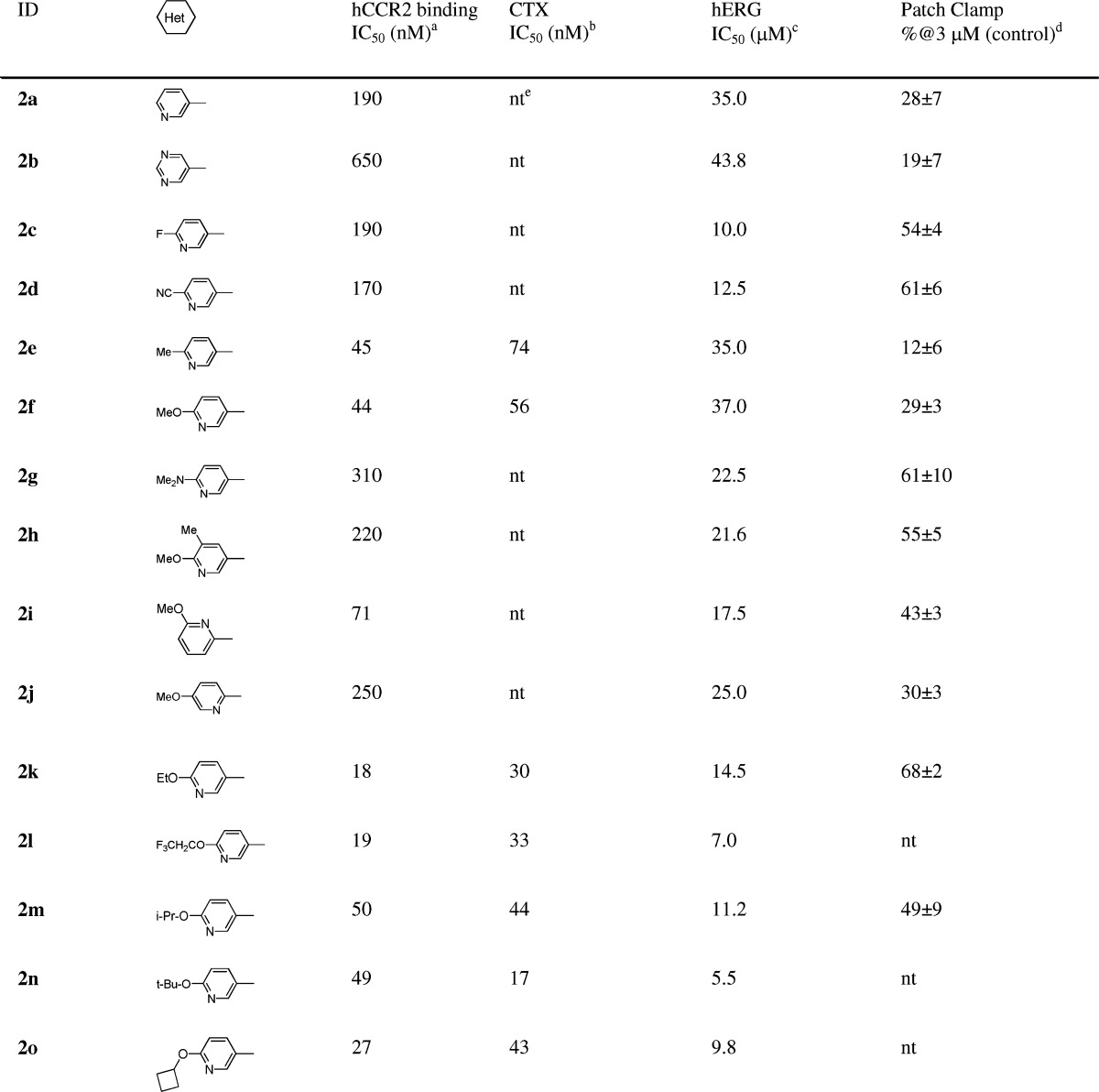
MCP-1 receptor binding assay in THP-1 cells: for IC50 > 100 nM (n = 1); for IC50 < 100 nM (n > 2, average values, SEM < ±25%).
MCP-1 induced chemotaxis in THP-1 cells.
hERG 3H-astemizole binding activity on the HEK-293 cell.
The membrane K+ current IKr in hERG-transfected HEK293 cells (n = 3).
nt: not tested.
Given the good in vitro selectivity between hCCR2 and hERG activities, 2e and 2f were selected for evaluation in an anesthetized guinea pig CV (GPCV) study. Disappointingly, both 2e and 2f still displayed electrocardiographic effects consisting of a dose dependent increase in ΔQTc. With a dosage of 10 mg/kg (i.v.), 2e and 2f exhibited +13% and +6% of the ΔQTc, respectively. These results led us to redefine our criteria on the in vitro hERG profile for advancing compounds into the GPCV study. Our efforts then focused on identifying compounds with decreased hERG affinity to our detection limit (IC50, 50 μM) while maintaining or improving hCCR2 activities. However, an attempt on determining the direct correlation between hERG binding and QTc prolongation could be challenging due to the impact on QTc from other factors, for example, serum protein binding. Therefore, the final selection of the compounds for further development should be solely determined by comprehensive in vivo CV safety evaluation.
Reviewing the recent literature led us to postulate that the weak basic pyridinyl group of 2e and 2f may participate in a cation−π interaction with the Tyr-652 residue of the hERG channel.18 Thus, elimination of the basic feature of the pyridinyl group would disrupt this interaction and further attenuate hERG binding. For the chemistry effort, we decided to examine the left-end aromatic ring with a series of five membered heterocyclic analogues. As illustrated in Table 2, substitution by a five membered heterocycle had a great impact on hCCR2 affinity. While incorporation of an oxazole (3), imidazole (4), pyrrozole (5), or thiadiazole (6) either efficiently attenuated or abolished hERG affinity, it also significantly reduced hCCR2 activity. To our satisfaction, installation of a 2-thiophene (7), 2-thiazole (8a), or 2-isothiazole (9) at the 1-postion of the cyclohexyl ring was tolerated for good hCCR2 binding affinity (IC50, 100, 62, 36 nM). Among them, 8a displayed relatively weak hERG binding affinity with an IC50 of 32 μM and promising separation between hCCR2 and hERG affinities, which prompted us to investigate this substitution pattern in detail.
Table 2. SAR of Five Membered Heterocyclic Analogues.
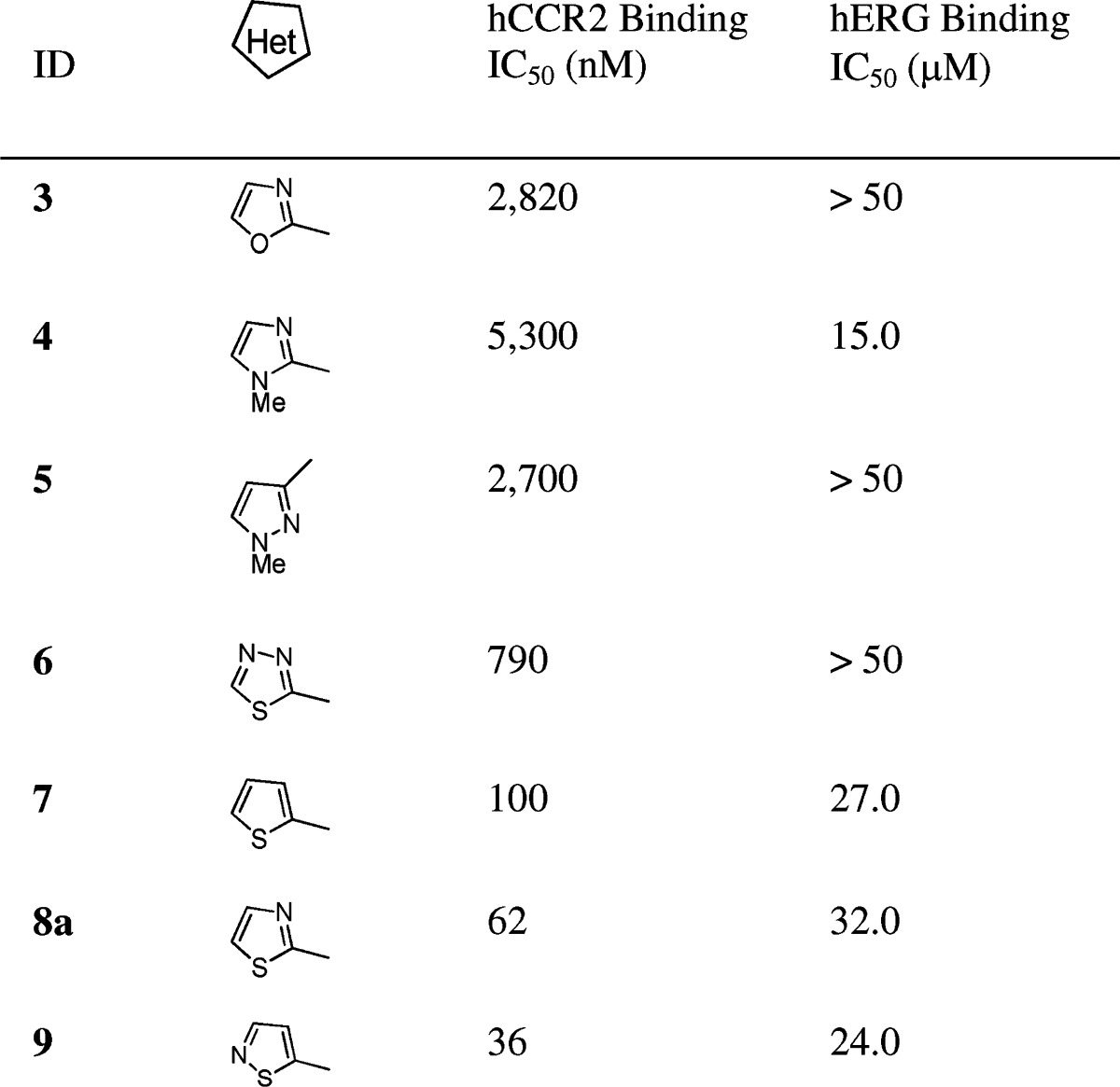
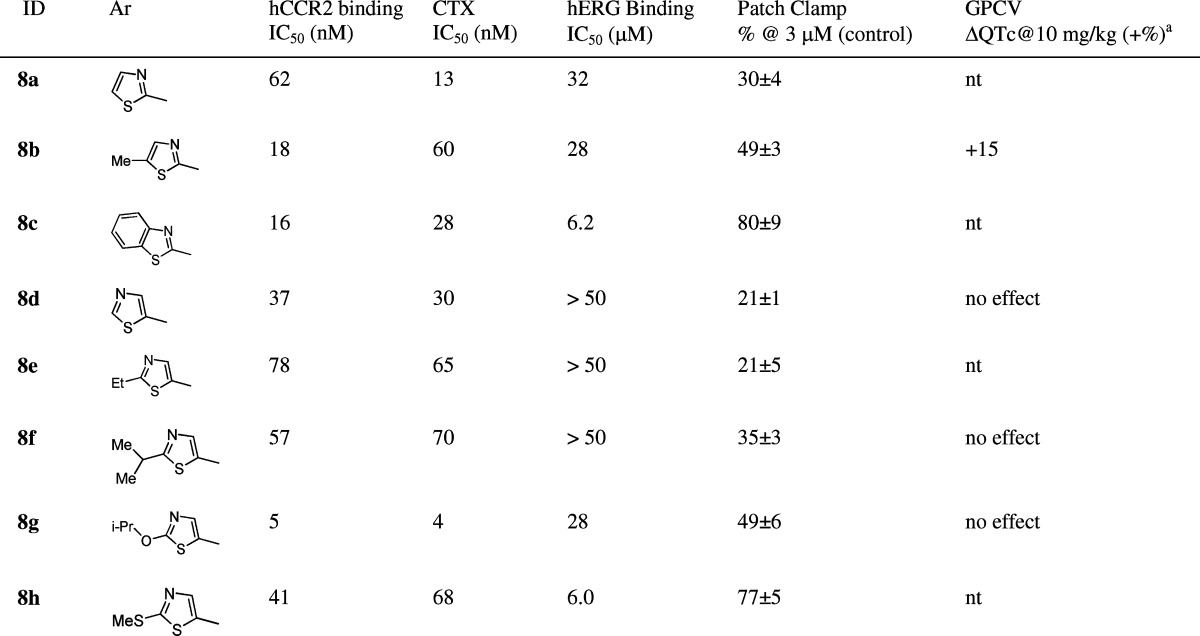
Table 3 highlighted the SAR within a series of thiazole substituted analogues. Installation of a thiazole moiety at the 1-position of the cyclohexyl ring was generally well tolerated for good hCCR2 activity. However, modification on the thiazole group had a great impact on the in vitro hERG profile and, hence, dominated the QTc effect in the GPCV study. There was a trend that more lipophilic thiazoles led to stronger hERG in vitro activity. Compared to 8a, simple methylation at the 3-position of the thiazole caused the blockade of hERG potassium current from 35% to 49% at 3 μM (8b). It is therefore not surprising that intravenous infusion of 8b in anesthetized guinea pigs triggered a dose dependent QTc prolongation (ΔQTc, +15% at 10 mg/kg). Compound 8c, bearing a hydrophobic benzothiazole moiety, possessed enhanced hERG in vitro activity. Installation of a 5-thaizole group onto the 1-position of the cyclohexyl ring (8d) remarkably attenuated the hERG binding affinity to greater than 50 μM. Consistent with its weak hERG affinity, 8d also displayed low inhibition on the potassium current in the patch clamp study (21% inhibition at 3 μM). To our delight, intravenous infusion of 8d in anesthetized guinea pigs had no effect on QTc up to 10 mg/kg. The plasma concentration reached 26 μM. Having identified the 5-thiazole group as the preferred left-end moiety, we extended our SAR studies to several substituted 5-thiazole analogues. Compounds 8e and 8f, with small alkyl substitutions, displayed good hCCR2 activities and the desired in vitro hERG profile. Furthermore, 8f demonstrated no effect on QTc up to 10 mg/kg (i.v.) in the GPCV study (plasma level, 10 μM). It is noteworthy that the 5-thiazole is not considered as a general pharmacophore for suppressing hERG in vitro activities and eliminating QTc prolongation in the GPCV study. As illustrated by 8g and 8h, while maintaining good hCCR2 activities, they both possessed enhanced inhibition on the potassium current (49% and 77% inhibition at 3 μM). While compound 8g displayed no effect on QTc up to 10 mg/kg (i.v.) in the GPCV study, the plasma concentration was only ∼3 μM which did not provide enough safety margin for further development. Here, an extremely narrow line appeared to determine the CV safety profile of this series.
Table 3. SAR of Thiazole Substituted Analogues.
Cumulative doses (0.1, 0.3, 1, 3, and 10 mg/kg) were administered incrementally as 5 min i.v. infusions at 0, 20, 40, 60, and 80 min. QTc intervals (ΔQTc) were measured and recorded at each time point as a percent change from baseline for each animal. Values are mean ± SEM (n = 3).
Given its superior in vitro hERG and GPCV safety profiles, 8d was selected to be further evaluated in an anesthetized dog CV safety study. Strikingly, it did not induce dose-dependent or notable effects on most cardiohemodynamic, functional respiratory, and electrophysiological parameters up to 10 mg/kg (i.v.) with the plasma level at 70 μM. It also did not induce changes in ECG morphology. Overall, 8d represented the first compound with a good CV safety profile as a development candidate.
Compound 8d was a selective hCCR2 inhibitor, showing no significant inhibitory activity at the concentration of 10 μM in Cerep and Invitrogen protein kinase panel screens. It did not significantly inhibit the binding of any of the relevant chemokines tested, including CCR1, CCR3, CCR4, CCR5, CXCR1, CXCR2, CXCR4, and CX3CR1 at the concentration of 25 μM.
Compound 8d also exhibited amendable DMPK profiles for further development. It was highly orally bioavailable and exhibited oral bioavailability of 70.2% in dogs when dosed at 6.7 mg/kg using 0.5% methocel as a vehicle (Cmax = 1617 ng/mL, AUClast = 5887 h·ng/mL). In addition, it showed 25.4% oral bioavailability in nonhuman primates (7.2 mg/kg p.o., Cmax = 740 ng/mL, AUClast = 3061 h·ng/mL) but only 19% of oral bioavailability in mice (10 mg/kg p.o., Cmax = 74 ng/mL, AUClast = 204 h·ng/mL), and 15.3% oral bioavailability in rats (10 mg/kg p.o., Cmax = 100 ng/mL, AUClast = 416 h·ng/mL).
Compound 8d only displayed a Ki of 9.6 μM for mCCR2 binding. Human CCR2 knock-in/murine CCR2 knockout mice (hCCR2KI) have been adopted to overcome the species issue and validate in vivo models. Compound 8d was assessed in several pharmacological models using hCCR2KI mice. In a thioglycollate-induced peritonitis (TG) model, it dose-dependently inhibited the influx of leukocytes, monocytes/macrophages, and T-lymphocytes into the peritoneal cavity with an ED50 of 3 mg/kg p.o. bid. In an OVA-induced asthma model, 8d inhibited airway eosinophil infiltration by 89% at a dose of 10 mg/kg p.o. bid. The efficacy of 8d in the mouse model suggests that it may have potential as a single agent in asthma to improve signs and symptoms and reduce airway resistance. The detailed pharmacological profile of 8d will be reported later.
The synthesis of this series is exemplified by compound 8d, as illustrated in Scheme 1. Thiazole 10 was treated with n-BuLi in THF at −78 °C, and the resulting anion was reacted with commercially available ketone 11 followed by desilylation with TBAF to obtain ketal 12 in 55% yield. Deprotection of the ketal group in 1 N HCl/acetone at room temperature gave the corresponding ketone 13 in 90% yield. Reductive amination of 13 with 14(16) using NaBH(OAc)3 gave a pair of cis–trans adducts as mixtures, which were separated by silica gel column chromatography to afford cis isomer (in terms of thiazole and azetidine moiety) 8d in 35% yield along with its trans isomer (structure not shown) in 40% yield.
Scheme 1. Synthesis of 8d.

Conditions: (a) n-BuLi, −78 °C. (b) TBAF, 2 steps, 55%. (c) HCl, acetone, room temperature, 90%. (d) NaBH(OAc)3, TEA, DCM, room temperature, 35% along with its isomer, 40%.
In conclusion, we have discovered a novel series of azetidinyl cyclohexanes as potent and selective hCCR2 antagonists. Through divergent SARs of hCCR2 and hERG on the left-end heterocyclic ring, 8d was identified as the lead compound. It exhibits potent hCCR2 activity, high selectivity, weak hERG affinity, and good oral bioavailability. Evaluated in guinea pigs and dogs, 8d possesses a clean CV safety profile and provides a good safety margin as a development candidate.
Acknowledgments
We thank LG Biology, CoE for Cardiovascular Safety Research, ADME/PK, and HOPS at JRD for technical assistance.
Supporting Information Available
Experimental procedures for the synthesis of 8d and characterization data for 2a–2o, 3–7, 8a–h, and 9, as well as in vitro and in vivo biological protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest..
Supplementary Material
References
- Charo I. F.; Ransohoff R. M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [DOI] [PubMed] [Google Scholar]
- Castellani M. L.; Bhattacharya K.; Tagen M.; Kempuraj D.; Perrella A.; Delutis M.; Boucher W.; Conti P.; Theoharides T. C.; Cerulli G.; Salini V.; Neri G. Anti-chemokine therapy for inflammatory diseases. Int. J. Immunopathol. Pharmacol. 2007, 20, 447–453. [DOI] [PubMed] [Google Scholar]
- Ogata H.; Takeya M.; Yoshimura T.; Takagi K.; Takahashi K. The role of monocyte chemoattractant protein-1 (MCP-1) in the pathogenesis of collagen-induced arthritis in rats. J. Pathol. 1997, 182, 106–114. [DOI] [PubMed] [Google Scholar]
- Fife B. T.; Huffnagle G. B.; Kuziel W. A.; Karpus W. J. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000, 192, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. K.; OH H. B.; Lee E. Y.; Lee J. E.; Kim Y. Y. Association between a genetic variation of CC chemokine receptor 2 and atopic asthma. Allergy 2007, 62, 207–208. [DOI] [PubMed] [Google Scholar]
- Dawson T. C.; Kuziel W. A.; Osahar T. A.; Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 1999, 143, 205–211. [DOI] [PubMed] [Google Scholar]
- Kanda H.; Tateya S.; Tamori Y.; Kotani K.; Hiasa K. I.; Kitazawa R.; Kitazawa S.; Miyachi H.; Maeda S.; Egashira K.; Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 2006, 116, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdi R.; Means T. K.; Ito T.; Smith R. N.; Najafian N.; Jurewicz M.; Tchipachvili V.; Charo I.; Auchincloss H. Jr.; Sayegh M. H.; Luster A. D. Differential role of CCR2 in islet and heart allograft rejection: tissue specificity of chemokine/chemokine receptor function in vivo. J. Immunol. 2004, 172, 767–775. [DOI] [PubMed] [Google Scholar]
- White F. A.; Feldman P.; Miller R. J. Chemokine signaling neuropathic pain. Mol. Interventions 2009, 9, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struthers M.; Pasternak A. CCR2 antagonists. Curr. Top. Med. Chem. 2010, 10, 1278–1298. [DOI] [PubMed] [Google Scholar]
- Hou C.; Sui Z.. CCR2 antagonists for the treatment of diseases associated with inflammation” Anti-inflammatory drug discovery; Levin J., Laufer S., Eds.; Royal Society of Chemistry, 2012; pp 350–390. [Google Scholar]
- Xue C.-B.; Wang A.; Meloni D.; Zhang K.; Kong L.; Feng H.; Glenn J.; Huang T.; Zhang Y.; Cao G.; Anand R.; Zheng C.; Xia M.; Han Q.; Robinson D. J.; Storace L.; Shao L.; Li M.; Brodmerkel C. M.; Covington M.; Scherle P.; Diamond S.; Yeleswaram S.; Vaddi K.; Newton R.; Hollis G.; Friedman S.; Metcalf B. Discovery of INCB3344, a potent, selective and orally bioavailable antagonist of human and murine CCR2. Bioorg. Med. Chem. Lett. 2010, 20, 7473–7478. [DOI] [PubMed] [Google Scholar]
- Xue C.-B.; Feng H.; Cao G.; Huang T.; Glenn J.; Anand R.; Meloni D.; Zhang K.; Kong L.; Wang A.; Zhang Y.; Zheng C.; Xia M.; Chen L.; Tanaka H.; Han Q.; Robinson D. J.; Modi D.; Storace L.; Shao L.; Sharief V.; Li M.; Galya L. G.; Covington M.; Scherle P.; Diamond S.; Emm T.; Yeleswaram S.; Contel N.; Vaddi K.; Newton R.; Hollis G.; Friedman S.; Metcalf B. Discovery of INCB3284, a potent, selective, and orally bioavailable hCCR2 antagonist. ACS Med. Chem. Lett. 2011, 2, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C.-B.; Wang A.; Han Q.; Zhang Y.; Cao G.; Feng H.; Huang T.; Zheng C.; Xia M.; Zhang K.; Kong L.; Glenn J.; Anand R.; Meloni D.; Robinson D. J.; Shao L.; Storace L.; Li M.; Hughes R. O.; Devraj R.; Morton P. A.; Rogier D. J.; Covington M.; Scherle P.; Diamond S.; Emm T.; Yeleswaram S.; Contel N.; Vaddi K.; Newton R.; Hollis G.; Metcalf B. Discovery of INCB8761/PF-4136309, a potent, selective, and orally bioavailable CCR2 antagonist. ACS Med. Chem. Lett. 2011, 2, 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanter J. C.; Markotan T. P.; Zhang X.; Subasinghe N.; Kang F.-A.; Hou C.; Singer M.; Opas E.; McKenney S.; Crysler C.; Johnson D.; Molloy C. J.; Sui Z. The discovery of novel cyclohexylamide CCR2 antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 7496–7501. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Hufnagel H.; Markotan T.; Lanter J.; Cai C.; Hou C.; Singer M.; Opas E.; McKenney S.; Crysler C.; Johnson D.; Sui Z. Overcoming hERG activity in the discovery of a series of 4-azetidinyl-1-arylcyclohexanes as CCR2 antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 5577–5582. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Hufnagel H.; Hou C.; Opas E.; McKenney S.; Crysler C.; O’Neill J.; Johnson D.; Sui Z. Design, synthesis and SAR of indazole and benzoisoxazole containing 4-azetidinyl-1-aryl-cyclohexanes as CCR2 antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 6042–6048. [DOI] [PubMed] [Google Scholar]
- Fernandez D.; Ghanta A.; Kauffman G. W.; Sanguinetti M. C. Physicochemical Features of the hERG Channel Drug Binding Site. J. Biol. Chem. 2004, 279, 10120–10127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.