

Abstract

A series of imidazo[1,2-a]pyridines which directly bind to HCV Non-Structural Protein 4B (NS4B) is described. This series demonstrates potent in vitro inhibition of HCV replication (EC50 < 10 nM), direct binding to purified NS4B protein (IC50 < 20 nM), and an HCV resistance pattern associated with NS4B (H94N/R, V105L/M, F98L) that are unique among reported HCV clinical assets, suggestive of the potential for additive or synergistic combination with other small molecule inhibitors of HCV replication.
Keywords: NS4B; hepatitis C virus; imidazo[1,2-a]pyridines; replicon
Hepatitis C virus (HCV) is a serious worldwide concern, infecting an estimated 170 million people (3% of the global population), including 4 million people in the United States.1,2 HCV leads to chronic liver disease, cirrhosis, and hepatocellular carcinoma, and it remains the most common indication for liver transplantation in developed countries. Between 8,000 and 10,000 deaths per year in the United States are attributed to complications of chronic HCV infection.1,2 The long-time standard of care, a combination of injected interferon and oral ribavirin, is poorly tolerated, contraindicated in a significant portion of the HCV-infected patient population, and affords a sustained virologic response in only half of those treated.1,2 The addition of one of the two recently approved protease inhibitors to the current standard of care significantly improves the overall cure rate; however, tolerability and drug–drug interactions remain a key concern.3,4
Identifying an oral drug cocktail that would both obviate the need for injected interferon and lead to sustained virologic responses over 90% remains the ultimate goal of most clinical development plans. The majority of clinical candidates currently undergoing investigation lead to HCV resistance arising in viral proteins, including well-characterized clinical mutations in the HCV protease, NS5A, and the HCV RNA-dependent RNA polymerase (NS5B).5,6 This article discloses the initial lead optimization of a series of potent HCV replication inhibitors structurally related to anguizole (1e),7,8 which is reported to directly bind to isolated HCV NS4B protein (Figure 1).8,9 These small molecules appear to act via a novel mechanism of action, which likely does not suffer from clinical cross-resistance to known NS5B, NS5A, or protease inhibitors currently approved or undergoing clinical trials and represents a potential option for combination therapy.
Figure 1.

Tritiated NS4B probes and anguizole.
a
Imidazo[1,2-]pyridines 1a–b are potent inhibitors of HCV in the in vitro replicon10 system, with measured EC50s for wild type (WT) genotype 1a and 1b replicons of <55 nM. Both 1a and 1b were recently disclosed in conjunction with other novel chemical matter optimized from a replicon-based HTS screen, with 1b having been identified following extensive optimization of 1a.11−15 Resistance passaging for compounds within this series (including 21 and anguizole7) revealed that key mutants arise in the HCV NS4B protein, specifically H94N/R, F98L, and V105L/M (genotype 1b). Imidazo[1,2-a]pyridines 1a–b show significant shifts in EC50s for replicons bearing these mutations (>20×). Titration of 1c with purified HCV NS4B protein affords a Kapp of 76 nM (data not shown) while an identical experiment with 1d affords a Kapp of 30 nM (Figure 2). Displacement of 1d with 1b affords a measured IC50 = 30 nM (Figure 3) as expected, and direct displacement of 1d by anguizole (1e) affords a measured IC50 = 22 nM. The affinities of additional analogues for purified NS4B protein (see Supporting Information) are reported using the direct displacement of 1d (see NS4B binding in subsequent tables16). These data are consistent with a direct interaction between small molecules within this class and the HCV NS4B protein.17
Figure 2.

Equilibirum binding of 1d for isolated His-Tagged NS4B. Kd = 30 nM (see Supporting Information).
Figure 3.

IC50 determination for 1b via 1d displacement from purified NS4B.
The development of initial lead molecule 1a was complicated by apparent thiophene-related, rapid in vitro/in vivo metabolism and irreversible trapping of glutathione in in vitro reactive metabolite assays. We set out to systematically optimize the DMPK and potency of 1a.
Versatile imidazo[1,2-a]pyridine carboxylic acid 4 could be prepared in four steps from commercially available 2-amino pyridine 2 (Scheme 1). Alkylation/cyclization with α-bromopyruvate, followed by selective chlorination with NCS and tandem Suzuki cross-coupling/hydrolysis, affords furan 4. Subsequent HATU mediated amide formation affords novel compounds 5–20. We found that a 3-substituted furan at C(5) and chlorine for bromine replacement at C(3) were well-tolerated relative to 1 (±6, Table 1), and we chose to focus first on the optimization of the amide moiety in the context of C(3) chloro and C(5) 3-furyl substituents.
Scheme 1. Synthesis of Core and Elaboration.

Conditions: (i) α-bromopyruvate, DMF, 50 °C (83%);11 (ii) NCS, DMF, 50 °C, 1 h, (91%);11 (iii) 3-furanylboronic acid, K3PO4, MeCN, Pd(dppf)Cl2, (82%);12 (iv) amine, HATU, DIPEA, DMF (20–85%).11−15
Table 1. Inhibition of HCV RNA in the Replicon Systema.
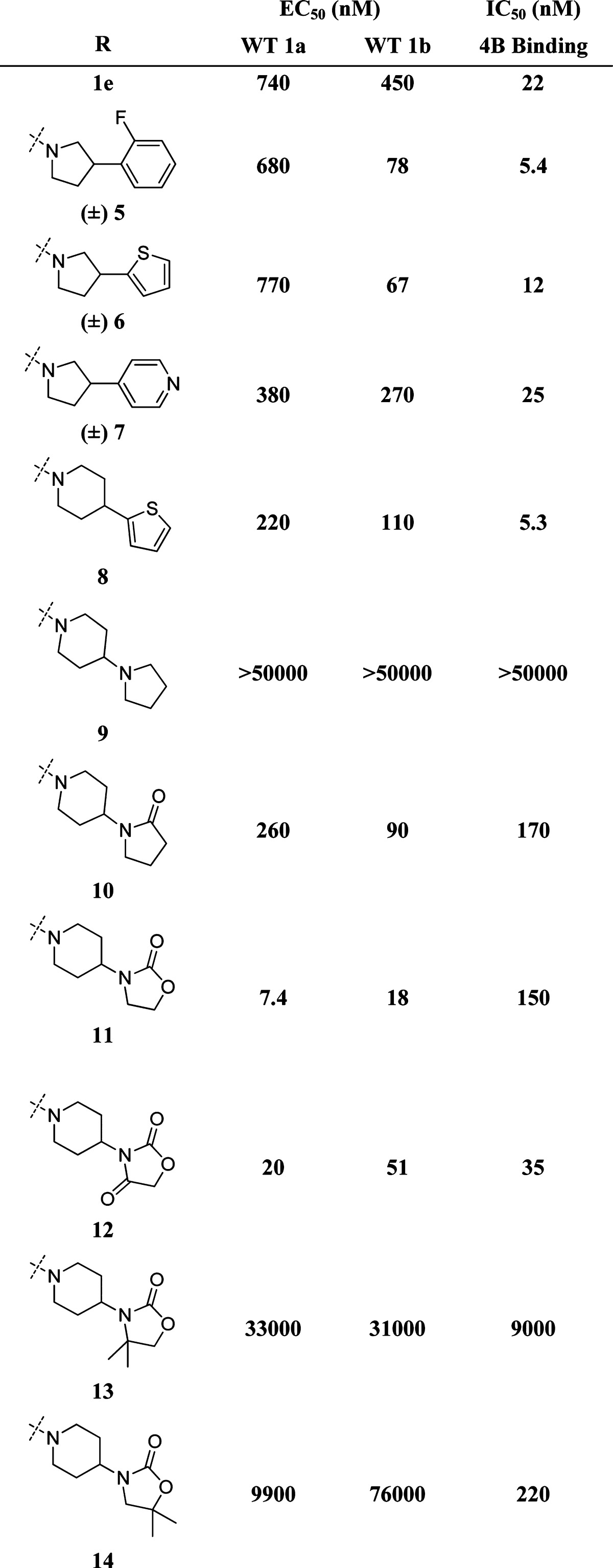
For 5–10, 12–14 WT 1a and 1b replicon data is the average of n = 3; for NS4B binding, data is the average of n = 6. For 11, WT 1a, 1b, and NS4B binding represent n = 506, 523, and 24, respectively. An analysis of variance (ANOVA) model was fit and used to link values to the compounds (see Supporting Information).
A range of 3-aryl (5) and 3-heteroaryl (6, 7) substituted pyrrolidines were well tolerated, giving submicromolar inhibitors of the HCV WT1a and WT1b replicons with retained ability to displace radioligand 1d from purified NS4B (Table 1). Ring expansion to the corresponding piperidine (i.e., 6 → 8) gave similarly equipotent HCV replication inhibitor 8.
Eliminating the chiral center proved most efficient for rapid assessment of novel amides. As such, we chose initially to further pursue the piperidine subseries. Basic pyrrolidine (9) exibited no HCV inhibition or NS4B binding, while incorporation of the corresponding cyclic amide (10) rescued both to a significant extent. Replacing the cyclic amide with the corresponding oxazolidinone or oxazolidine dione afforded 11 and 12, respectively. Oxazolidinone 11 was 5–10 times more potent than the corresponding amide (10). Direct substitution of the oxazolidinone ring was not well tolerated (see 13 and 14). In fact, the size, substitution, and electronics of the oxazolidinone ring appeared critical, as dozens of ring-opened and ring-substituted oxazolidinone analogues showed a much reduced ability to inhibit HCV replication in vitro and/or to bind directly to purified NS4B.11−15
With potent oxazolidinone 11 and oxazolidine dione 12 in hand, we examined standard DMPK properties, including P450 inhibition, reactive metabolite potential, and in vivo rat PK. We found 11 had no significant P450 liabilities or reactive metabolite potential in a glutathione trapping assay, but exhibited rapid in vivo clearance in rat (Table 2). Importantly, high circulating levels of 12 were observed following IV or PO dosing of 11. Although dione 12 represented a marginal improvement in in vivo clearance, potency, and overall exposure relative to 11, it was positive in our assessments of reactive metabolite. With these data in hand, we set out to identify a suitable oxazolidinone replacement, which could not be oxidized in vivo to reactive oxazolidine diones such as 12.
Table 2. Rat DMPK Profile for 11 and 12a.
| 11 | 12 | |
|---|---|---|
| clearance | 72 | 46 |
| Vdss | 2.5 | 3.1 |
| PO T1/2 | 1.9 | 3 |
| AUC0–24 | 700 | 1100 |
| %F | 60% | 60% |
Dosed as a solution in DMSO/4% HPβCD in acetic acid (5:95); IV dosed 1 mg/kg; PO dosed 5 mg/kg; clearance (mL/min/kg); Vdss (L/kg); T1/2 (h), AUC (ng·h/mL). All n = 3 animals.
Recognizing that the carbonyl moiety was an important pharmacophore (i.e., 9 → 10, Table 1), we envisioned a number of modifications which might position this critical group in similar space to 10. Methyl piperazinone 15 exhibited a 1.2 μM IC50 in our radioligand displacement assay, but aryl 16 showed more promising potency, with the addition of the pendant ring affording 50-fold improvements in NS4B affinity and replicon potency. In contrast to the oxazolidinone series, incorporation of a properly placed carbonyl gave a less dramatic (i.e., 17 → 16) change in replicon potency and NS4B binding affinity. However, further refinement to cyclohexyl afforded potent HCV replicon inhibitor 18.
Bare cyclohexyl 18 suffered from rapid in vitro and in vivo clearance in rat (data not shown, Clrat ≫ hepatic blood flow). Metabolite ID studies with rat/human hepatocytes indicated a principal route of metabolism of 18 was via direct oxidation of the pendant cyclohexyl. Preoxidation of the cyclohexyl was tolerated with respect to NS4B binding and replicon potency, with a trans relative configuration of a pendant hydroxyl better tolerated than the corresponding cis arrangement (20 vs 19, Table 3).
Table 3. Inhibition of HCV RNA in the Replicon Systema.
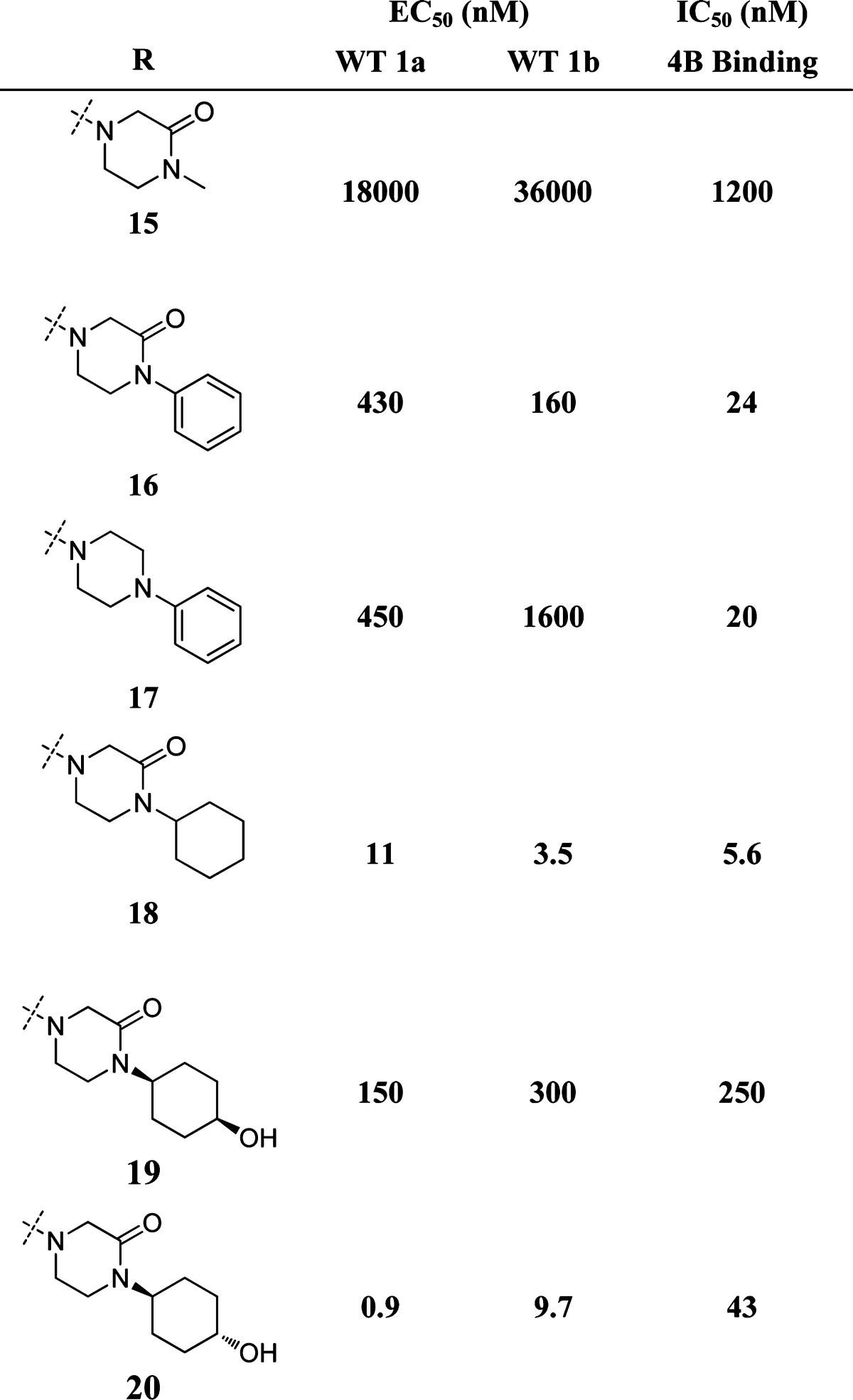
For WT 1a and 1b replicon data is the average of n = 3; for NS4B binding, data is the average of n = 6. XC50 determinations performed as for Table 1.
The imidazopyridine analogues, in general, display a reasonable correlation (R2 = 0.48) between direct NS4B binding and cell-based replicon inhibition (see Supporting Information Table SI-4). While permeability for analogues in this series was uniformly high, many of the outliers with reduced potency in the replicon assays (e.g., 5 and 6) relative to the NS4B binding assay were found to have poor measured solubility (<15 μM). Precipitation of compound at higher concentrations likely confounds EC50 interpretation and reproducibility. Conversely, analogues lacking an extra aromatic ring (9–15, 18–21) afforded maximum measured solubilities of >500 μM, were generally well-behaved in the replicon assay, and as such constituted the focus of our efforts.
Overall hydroxycyclohexyl moieties 19 and 20 gave an improved in vivo DMPK profile (data not shown) relative to 18. Despite reductions in in vivo clearance, 20 suffered from positive reactive metabolite, apparently now as a result of oxidation of the pendant furan (not observed directly for any previous molecules). A brief survey of less reactive functionality at the furan position revealed that small, alkyl groups in addition to heteroaryl functionality were well tolerated. This survey led to the subsequent identification of 21, wherein the pendant furan was replaced by a cyclopropyl. The same trends in SAR observed with the furan functionality were observed in the context of the cyclopropyl as well. For example, the cis isomer corresponding to 21 shows significant reductions in both NS4B binding and replicon inhibition (see Table SI-3, Supporting Information).
Compound 21 was equipotent to furan 20 and showed no shift in potency in well-characterized replicon mutants arising in NS5B, NS3, or NS5A (Table 4), consistent with a novel MOA involving direct interaction with NS4B. In contrast, stable replicons bearing NS4B mutations H94N or V105M showed significant resistance to 21 (>100×; see Table 4), while transiently transfected NS4B mutations F98L, H94R, and V105L showed resistance as well. Further, 21 possessed no measurable P450 (3A4, 2C9, 2D6, 1A2, 2C19) or reactive metabolite liabilities. In vivo profiling of 21 in rat revealed improved clearance and oral exposure relative to 11, 18, and 20 when dosed in rat or dog at 5 mg/kg (Table 5).
Table 4. Replicon Profile for 21a.
| mutant replicon | gene | EC50 (nM) |
|---|---|---|
| NS4B binding | 60 | |
| WT 1a | 0.81 | |
| WT 1b | 5.4 | |
| 1B H94N | NS4B | 200 |
| 1B V105M | NS4B | 800 |
| 1B H94R* | NS4B | 53 |
| 1B V105L* | NS4B | 38 |
| 1B F98L* | NS4B | 27 |
| 1B A156T* | NS3 | 1.0 |
| 1B M414T* | NS5B | 1.0 |
| 1B L31V* | NS5A | 1.0 |
WT 1a and WT 1b data represents n = 137 and 138, respectively; for NS4B binding, data represents n = 6. XC50 determinations performed as for Table 1. For 1B replicons H94N (n = 65), for 1B V105M (n = 59), for 1B A156T/M414T/L31 V (n = 2), H94R (n = 30), F98L (n = 20), and V105L (n = 182). * denotes transiently transfected replicons; all other replicons were stable.
Table 5. Rat and Dog DMPK Profile for 21.
Dosed as a solution in DMSO/Solutol/20% HPβCD (10:10:80); IV dosed 1 mg/kg; PO dosed 5 mg/kg; clearance (mL/min/kg); Vdss (L/kg); T1/2 (h), AUC (ng·h/mL).
Imidazo[1,2-a]pyridine (21) inhibits HCV replication in vitro with EC50 < 10 nM for genotype 1a and 1b replicons (Table 4), demonstrates good oral exposure in rodent and dog (Table 5), and maps its resistance to HCV NS4B (Table 4), unique among existing HCV inhibitors undergoing clinical trials. These properties make this class of imidazo[1,2-a]pyridines a promising and unique class of HCV replication inhibitors. We will report the full in vivo characterization of 21 and other related development candidates in due course.
Acknowledgments
We thank Steven Novick for the statistical analyses described in this communication and the Supporting Information. The authors acknowledge Randy Bledsoe and Tom Consler for the purification of the NS4B protein and the antibody preparation. The authors acknowledge Todd Baughman for conducting the reactive metabolite assays. The authors acknowledge Luz Helena Carballo for replicon screening.
Supporting Information Available
Replicon and NS4B binding assay protocols, synthetic procedures, compound purities, protein purification protocols, in vivo/in vitro DMPK protocols, details of radioligand syntheses, and statistical methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): All authors are current or former employees of GlaxoSmithKline.
Author Present Address
§ Kanion USA, Inc., 3916 Trust Way, Hayward, CA 94545.
Author Present Address
∥ Alios BioPharma, 260 East Grand Ave., 2nd Floor, South San Francisco, CA 94080.
Author Present Address
⊥ Gilead Sciences, Department of Structural Chemistry, 333 Lakeside Drive, Foster City, CA 94404.
Supplementary Material
References
- Kim A.; Timm J. Resistance mechanisms in HCV: from evolution to intervention. Expert Rev. Anti-Infect. Ther. 2008, 64463–478. [DOI] [PubMed] [Google Scholar]
- Almasio P.; Ingrassia D.; Vergara B.; Filosto S. New therapeutic prospects in HCV treatment. Expert Rev. Anti-Infect. Ther. 2008, 66775–779. [DOI] [PubMed] [Google Scholar]
- Kwong A.; Kauffman R.; Hurter P.; Mueller P. Discovery and development of telaprevir: an NS3–4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol. 2011, 2911993–1003. [DOI] [PubMed] [Google Scholar]
- Chen K.; Njoroge F. The Journey to the Discovery of Boceprevir: An NS3-NS4 HCV Protease Inhibitor for the Treatment of Chronic Hepatitis C. Prog. Med. Chem. 2010, 49, 1–36. [DOI] [PubMed] [Google Scholar]
- Soriano V.; Vispo E.; Poveda E.; Labarga P.; Martin-Carbonero L.; Fernandez-Montero J.; Barreiro P. Directly acting antivirals against hepatitis C virus. J. Antimicrob. Chemother. 2011, 6681673–86. [DOI] [PubMed] [Google Scholar]
- Pockros P. Drugs in development for chronic hepatitis C. Expert Opin. Biol. Ther. 2011, 11121611–22. [DOI] [PubMed] [Google Scholar]
- Bryson P.; Cho N.; Einav S.; Choongho L.; Tai V.; Bechtel J.; Sivaraja M.; Roberts C.; Schmitz U.; Glenn J. A small molecule inhibits HCV replication and alters NS4B’s subcellular distribution. Antiviral Res. 2010, 8711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chunduru S.; Benetatos C.; Nitz T.; Bailey T.; Chunduru S.; Benetatos C.; Nitz T.; Bailey T.; Benatatos C. A.. Composition useful in the prophylaxis or treatment of viral infection e.g. hepatitis C infection in living host comprises amides or N and/or S containing heterocyclic compounds and carrier patent. WO2005051318, 2005.
- The potential for this novel route of HCV inhibition has been reviewed elsewhere:Rai R.; Deval J. New opportunities in anti-hepatitis C virus drug discovery: Targeting NS4B. Antiviral Res. 2011, 90293–101. [DOI] [PubMed] [Google Scholar]
- Pietschmann T.; Lohmann V.; Kaul A.; Krieger N.; Rinck G.; Rutter G.; Strand D.; Bartenschlager R. Persistent and Transient Replication of Full-Length Hepatitis C Virus Genomes in Cell Culture. J.Virology 2002, 76, 4008–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banka A.; Catalano J. G.; Chong P. Y.; Fang J.; Garrido D. M.; Peat A. J.; Price D. J.; Shotwell J. B.; Tai V.; Zhang H.. Preparation of piperazinyl antiviral agents. WO 2011041713, 2011.
- Baskaran S.; Maung J.; Neitzel M. L.; Rai R.; Slododov I.; Tai V. t. W.-F. Preparation of imidazopyridine derivatives for treating viral infections. US 20100204265, 2010.
- Baskaran S.; Maung J.; Neitzel M.; Rai R.; Slobodov I.; Tai V.. Preparation of imidazopyridine derivatives for treating viral infections . WO 2010091409, 2010.
- Banka A.; Baskaran S.; Catalano J.; Chong P.; Dickson H.; Fang J.; Maung J.; Neitzel M. L.; Peat A.; Price D.; et al. Preparation of piperidinyl cyclic amido compounds as HCV antiviral agents. WO 2010091411, 2010.
- Schmitz F. U.; Tai V.; Rai R.; Roberts C.; Abadi A. D. M.; Baskaran S.; Slobodov I.; Maung J.; Neitzel M. L.. Preparation of substituted imidazopyridine derivatives and analogs for use as antiviral agents. WO 2009023179, 2009.
- Much early optimization was carried out using a binding assay incorporating 1c. Improved ligand stability of 1d relative to 1c led us to transition to the assay reported herein following the discovery of 1b.
- Visualization of NS4B membrane associated foci was attempted to investigate compound mechanism of action; however, changes in NS4B localization upon compound dosing were not observed. A membrane interaction assay described in ref (7) was also reproduced but did not show a correlation between assay activity and compound potency in replicon or binding assays. A full biological characterization of compound 21 will be published separately.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.