

Abstract

Inhibitors of the cancer-related cysteine isopeptidase human ubiquitin-specific proteases 7 (USP7) and 47 (USP47) are considered to have potential as cancer therapeutics, owing to their ability to stabilize the tumor suppressor p53 and to decrease DNA polymerase β (Polβ), both of which are potential anticancer effects. A new class of dual small molecule inhibitors of these enzymes has been discovered. Compound 1, a selective inhibitor of USP7 and USP47 with moderate potency, demonstrates inhibition of USP7 in cells and induces elevated p53 and apoptosis in cancer cell lines. Compound 1 has been shown to demonstrate modest activity in human xenograft multiple myeloma and B-cell leukemia in vivo models. This activity may be the result of dual inhibition of USP7 and USP47. To address issues regarding potency and developability, analogues of compound 1 have been synthesized and tested, leading to improvements in potency, solubility, and metabolic reactivity profile. Further optimization is expected to yield preclinical candidates and, ultimately, clinical candidates for the treatment of multiple myeloma, prostate cancer, and other cancers.
Keywords: Ubiquitin isopeptidase, deubiquitylase, p53, Polβ
Ubiquitin plays a key role in the regulation of the cell content of certain proteins, including some of those that are important in diverse diseases including cancer, muscle wasting, and inflammation.1 Ubiquitin can be conjugated to the ε-amino group of lysine residues by E3 ligases.2 Polyubiquitylated proteins are typically degraded by the proteasome. Prior to engagement by the proteasome, however, ubiquitin can be removed from proteins by deubiquitylating enzymes (DUBs), thus sparing the proteins from degradation by the proteasome. Approximately 80 functional DUBs have been identified and have been divided into five classes, four of which are cysteine proteases.3 The largest class comprising more than 50 enzymes are the ubiquitin specific proteases (USP/UBP).4
We have been interested in inhibiting USP7, a cysteine protease associated with prostate cancer and other refractory solid malignancies.5 USP7 preferentially deubiquitylates HDM2, which is a ubiquitin E3 ligase for the tumor suppressor p53,4,5 thereby preventing the degradation of HDM2 in the proteasome. USP7 mediated stabilization of HDM2 results in a reduction of the cellular p53 content and may protect damaged cells from apoptotic death.5,6 Inhibition of USP7 reverses this process, increasing levels of the tumor suppressor p53, which promotes cell cycle arrest and apoptosis of cancer cells. Recently, additional substrates of USP7 have been reported, including claspin, FOXO4, and PTEN. Many of these substrates occupy key signaling nodes in cancer cells, highlighting the importance of USP7 as an antineoplastic target, independent of p53 status.5
Phylogenetic analysis revealed that USP47 is one of the DUBs most closely related to USP7,7 and recent data support the observation that USP47 is also an emerging oncology target. For example, USP47 regulates DNA base excision repair (BER) by controlling deubiquitylation (and thus the cellular content) of Polβ, which acts as both a DNA polymerase and lyase in BER.8 It has been shown that knockdown of USP47 leads to reduced levels of Polβ, defective DNA BER, and decreased cell viability. Knockdown of USP47 reduces tumor cell proliferation and enhances the cytotoxic activity of chemotherapeutic agents.9
Previously, HBX 41,108 (7-chloro-9-oxo-9H-indeno(1,2b)pyrazine-2,3-carbonitrile, CAS No. 924296-39-9) was reported to inhibit purified USP7 as well as USP7 activity in cells.10 While HBX 41,108 is selective against many other families of proteases, it also inhibits caspase 3 and multiple DUBs, including USP5 and USP8.11 Our work, which has resulted in the discovery of a new family of selective USP7/USP47 inhibitors with potential synergistic effects for the treatment of cancer, began by screening commercial libraries for USP7 inhibitors using a Ub-PLA2 (Ub-CHOP) reporter assay.12 Because the assay does not distinguish between inhibition of USP7 and inhibition of the reporter enzyme PLA2, all active compounds were counter-screened for inhibition of PLA2 and only those negative in this assay accepted as USP7 inhibitors. Several compounds were identified as inhibitors active in the low micromolar range, and additional testing pointed to P005091 (1; Figure 1) as a suitable starting point for further medicinal chemistry optimization. Compound 1 is selective; it exhibited little or no inhibition activity for many USP variants or other classes of protease, including caspase, cathepsins, calpain, and metalloproteases and serine proteases. Of the proteases tested, only USP47 was as susceptible as USP7 to inhibition by compound 1. This compound accelerated the degradation of the USP7 substrate HDM2 in several cancer cell lines, inhibited the growth of HCT-116 human colorectal cancer cells with an EC50 of 11 μM, and inhibited cancer cell growth synergistically with doxorubicin, etoposide, or mechlorethamine (Supporting Information Figure 1 and Kumar, unpublished results). It significantly increased the lifespan and reduced the tumor burden of mice in human multiple myeloma and B cell leukemia xenograft models [Chauhan et al., in press].
Figure 1.
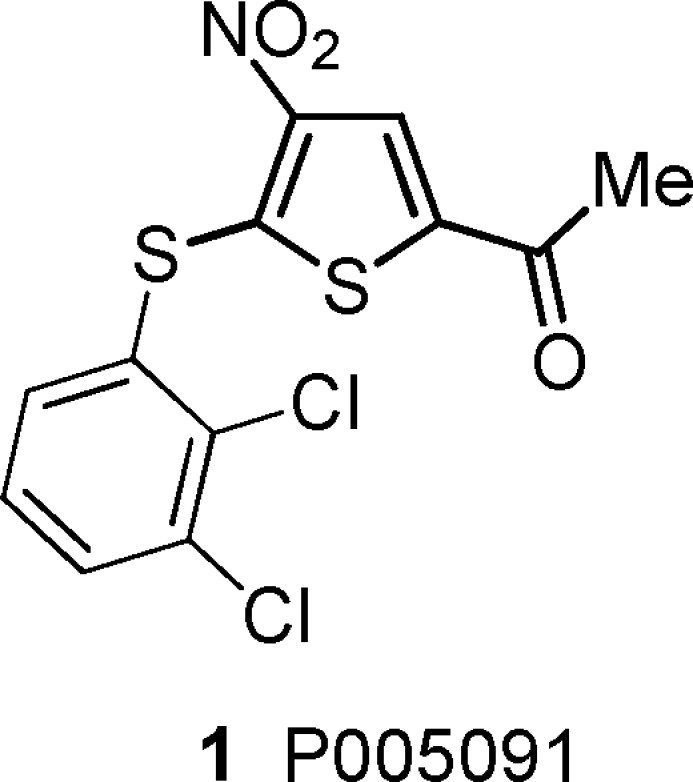
Structure of the USP7 inhibitor lead P005091 {1-[5-(2,3-dichlorophenylsulfanyl)-4-nitro-2-thienyl]ethanone}.
Characterization of 1 in biological matrixes was limited, as it gave a very low MS signal at biologically relevant concentrations. Some of the initially prepared analogues were detectable at relevant concentrations, but they exhibited low recovery from glutathione (GSH) solutions and poor plasma stability. Thus, the goals of the work reported here included identifying compounds with improved USP7 inhibition, retention of selectivity, increased solubility, increased stability in GSH solutions, and improved plasma stability.
A series of compounds were prepared to explore SAR around the lead compound (1). Initial efforts focused on the thiol substitution at position 5. To this end, compounds 1–6 were prepared as illustrated in Scheme 1, in which various substitutions on the phenyl ring were explored. Subsequently, we focused on substitution at the 2 position of the thiophene ring. It was anticipated that substitution at this position would provide an excellent opportunity for introducing aqueous solubility into the molecule. Thus, compounds 7–11 were prepared as shown in Scheme 2.
Scheme 1. Synthesis of 2-Acetyl-4-nitro-5-arylsulfanylthiophenes.

Scheme 2. Synthesis of Thiophene-2-carboxamides.

Realizing that the nitro group could contribute to reactivity by way of thiol displacement, we prepared a series of potentially less reactive cyano compounds. Initially, 1-[4-cyano-5-(2,4-dichlorophenyl)sulfanyl-2-thienyl]ethanone (12) was prepared from 3 by reduction of the nitro group to the amine 19 followed by Sandmeyer conversion to the bromide 20, and, finally, copper catalyzed displacement of the bromide by cyanide. Attempts to follow this procedure for the preparation of 13 and 14 resulted in poor yields. In a modified approach (Scheme 3), the 3-amino cyano ester 21 was obtained in one step from reaction of methyl thioglycolate with 2-[bis(methylthio)methylene] malononitrile.13 Diazotization of the amino group followed by oxidation provided the sulfone 22. Reaction of 22 with 3,5-dichloropyridyl-5-thiol gave 23, which was converted to analogues 13 and 14 via reaction with trimethylaluminum activated amines.14
Scheme 3. Synthesis of Thiophene-4-nitriles.

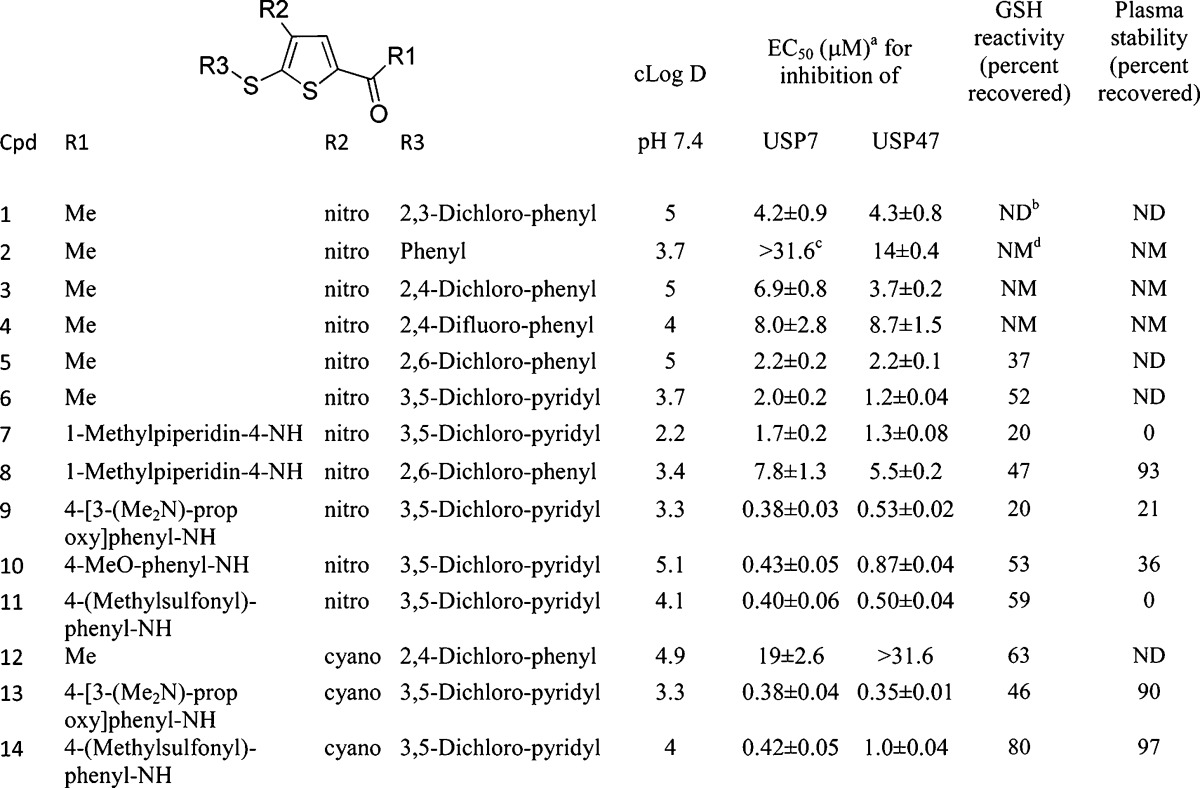
A summary of the biological activity and properties of compounds 1–14 is provided in Table 1. SAR data on additional compounds are presented in Supporting Information Tables 1–5. Although 2, the unsubstituted phenylsulfanyl analogue of 1, displayed limited activity against USP7, the dihalo analogues (3–5) demonstrated activity similar to that of 1. Compound 5 proved to be the most potent of these analogues, being twice as active as 1. Compound 4, the difluoro analogue of 3, was similar in activity to analogue 3. Furthermore, the inhibitory activity of compound 4 (P22077) vs USP7 and USP47 in human cells was established and is provided in a recent publication from our group.15 Of particular note, the 3,5-dichloropyridyl analogue 6 was approximately 2.5 times more potent as a USP7 inhibitor than analogue 1.
Table 1. Characterization of Compound 1 and Analogues.
n = 3–50.
Not detectable.
The EC50 value was >31.6 μM.
Not measured.
Carboxamide analogues were selected for further study because they allowed convenient introduction of diverse substituents as well as the opportunity to increase aqueous solubility. Compound 7 inhibited USP7 activity with a potency similar to that of 6 and exhibited a cLogD value 1.5-fold lower than that of compound 6. Compound 7, however, proved to be reactive toward GSH and unstable in plasma. The GSH reactivity may be related to the basic amine moiety which may deprotonate the GSH thiol group and thus activate it as a nucleophile. The nature of the reactivity of compound 7 with GSH was investigated further, and LC-MS analysis revealed the presence of an adduct with a mass of 575.15, which corresponds to the glutathione thiol displaced 5-sulfide group of compound 7 (Supporting Information Figure 2).
Since the 4-nitro group of compounds 1–11 is a strong activator for the nucleophilic displacement of the 5-thia moiety, we explored a wide variety of replacements for the 4-nitro group, as shown in Supporting Information Tables 2 and 5. The cyano group was identified as the only replacement group of those studied that retained reasonable USP7 activity. Compound 12 was approximately three times less potent than the related nitro compound 3. However, compound 13 inhibited USP7 with potency similar to that of 9 and demonstrated good recovery from exposure to plasma. Despite these improved properties, compound 13 remained reactive against GSH. Compound 14, the 4-cyano analogue of 11, had equivalent potency against USP7 and, in addition, demonstrated good stability vs GSH and acceptable recovery from plasma. Consistent with other compounds in this series, compound 14 is a selective USP7 (EC50 = 0.42 μM)/USP47 (EC50 = 1.0 μM) inhibitor but does not inhibit caspase 3, calpain 1, 20S proteasome, and a panel of representative USPs (USP2, USP5, USP8, USP21, and USP28; EC50 > 31.6 μM; Supporting Information Table 6). It is important to note that compounds 1–14 all exhibited negligible inhibition of the unrelated cysteine protease, caspase 3, illustrating the selective nature of these USP7/USP47 inhibitors (data not shown).
To summarize the SAR, we have found that substituting a 5-pyridyl thiol group for a 5-phenyl thiol group in the original lead compound 1 results in compounds with increased potency. Further, the optimal activity was observed with halo substituted aromatic rings at position 5 (compare 1 and 2). In addition, carboxyamides at C2 provide an opportunity to prepare compounds with enhanced aqueous solubility while maintaining activity vs USP7 and USP47. Finally, initial data suggest that substitution of a cyano for a nitro group on the thiophene ring results in enhanced stability, as illustrated by reduced GSH reactivity and excellent plasma stability (compare 11 and 14). Further investigation of this observation will be the subject of future exploration.
Compounds 1 and 14 were compared directly for their ability to inhibit the proliferation of human colon cancer, HCT-116 cells. Data presented in Supporting Information Figure 1 illustrate that, relative to compound 1, compound 14 exhibits enhanced potency against HCT-116 cells. An important property of a therapeutic inhibitor of cellular enzymes such as USP7 is the ability to modulate appropriate cellular pharmacodynamic markers such as the HDM2 substrate p53. Inhibition of USP7 destabilizes HDM2, resulting in an increase in p53 and its downstream target p21.5 This property was investigated using cells from the colon cancer line HCT-116; the cells were exposed to 30 μM 14, and the cell lysates were assayed by immunoblotting for p53 and p21. Figure 2 shows a comparison of the treated and control cells. Consistent with prior reports, DMSO induced a minor increase in p21 protein and there was no change in p53 protein.16,17 In contrast, treatment of cells with compound 14 resulted in an increase in p53 and an appreciable induction of p21 protein over the DMSO control. Thus, compound 14 penetrated the cells and modulated p53 and p21 as expected for a USP7 inhibitor. In agreement with the dual USP7/USP47 targeting hypothesis, compound 14 modestly accelerated the degradation of polβ protein in HeLa cells (data not shown).
Figure 2.
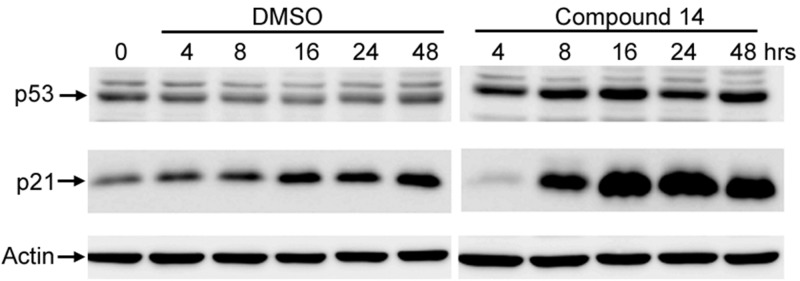
Cellular activity of compound 14. HCT-116 human colon cancer derived cells were incubated with vehicle (0.3% DMSO) or 30 μM compound 14, and lysates were prepared and subjected to SDS-PAGE and Western blotting analysis. Shown are relevant portions of immunoblots probed with antibodies to (top) p53; (middle) p21; and (bottom) actin (loading standard).
The comparable inhibitory activity toward USP7 and USP47 shown by the compounds in this series in Table 1 is not surprising given that a phylogenetic comparison of the core catalytic domains of the USP family reveals that USP47 is one of the DUBs most closely related to USP7.7 The parallel inhibition noted has biological significance, as USP7 and USP47 both play important roles in cell survival, but by different mechanisms.5,6,8,9
USP7 exerts anti-apoptotic effects by stabilizing HDM2, thereby destabilizing p53, and also exerts a prosurvival effect by stabilizing the DNA damage repair protein claspin. In addition to HDM2, other USP7 substrates have been reported, many of which function in diverse neoplastic pathways. Thus, a USP7 inhibitor would exert pro-apoptotic, antigrowth effects in p53 wild type and p53 mutant tumors.5
USP47 is an emerging anticancer target, as it regulates base excision repair via Polβ and also modulates cell growth and augments the activity of chemotherapeutic agents. Among the sequelae of stress induced by radiation, mutagens, or other agents are DNA strand breaks, which are lethal to cells unless they are repaired. Due to compromised DNA damage repair, many tumors are highly sensitive to inhibition of DNA repair enzymes such as Polβ. The cellular level of Polβ is regulated by a balance between ubiquitylation by E3 ligases decreasing Polβ concentration and deubiquitylation by USP47, increasing it. Consistent with this model, treatment of cancer cell lines with compound 1 or 7 sensitizes them to numerous genotoxic agents (Kumar, unpublished results).
It is important to note that a dual inhibitor (such as USP7/USP47), in comparison to compounds that inhibit only one pathway, likely would have increased single agent or adjuvant (with genotoxic agents) anticancer efficacy and reduced susceptibility to the development of drug resistance. We have identified deubiquitylating enzyme inhibitors that are among the most selective described so far for these DUBs; some have sub-micromolar potency as inhibitors of USP7 and USP47. We discovered that members of this series are inhibitors of USP7 and of USP47 that may act synergistically. In addition, we have identified the 3,5-dichloropyridine moiety as a potent USP7 pharmacophore. Work is continuing in this area to identify potential clinical candidates for the treatment of certain cancers.
Acknowledgments
We thank Ms. Danielle Fagnani for technical assistance.
Glossary
Abbreviations
- BER
base excision repair
- DUB
deubiquitylating enzyme
- Polβ
DNA polymerase β
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- mCPBA
3-chloroperoxybenzoic acid
Supporting Information Available
Additional SAR tables, synthetic procedures, compound characterization, biological assay data, and bioassay methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): J.W., P.C., J.L.M., M.P.K., D.M.M., K.G.S.K., S.J.G., M.R.M., and B.N. are or were employees of Progenra, Inc. when this work was performed. Joseph Weinstock and William Kingsbury are paid consultants of Progenra, Inc.
Supplementary Material
References
- Dye B.; Schulman B. A. Structural mechanisms underlying posttranslational modification by ubiquitin-like proteins. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 131–150. [DOI] [PubMed] [Google Scholar]
- Hershko A.; Ciechanover A. The ubiquitin-proteasome pathway. Annu. Rev. Biochem. 1998, 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Nijman S. M.; Luna-Vargas M. P.; Velds A.; Brummelkamp T. R.; Dirac A. M.; et al. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu F. E.; Ventii K. H.; Wilkinson K. D. Regulation and cellular roles of ubiquitin-specific deubiquitibnating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson B.; Suresh Kumar K. G. The multifaceted roles of USP7: new therapeutic opportunities. Cell. Biochem. Biophys. 2011, 60, 61–68. [DOI] [PubMed] [Google Scholar]
- Waring D.; Lebman J. A.; Batuello C. N.; Mayo L. D. Controlling the Mdm2-Mdmx-p53 circuit. Pharmaceuticals 2010, 3, 1576–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catic A.; Fiebiger E.; Korbel G. A.; Blom D.; Galardy P. J.; et al. Screen for ISG15-crossreactive deubiquitinases. PLoS ONE 2007, 2, e679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. L.; Khoronenkova S. V.; Edelmann M. J.; Kessler B. M.; Dianov G. L. USP47 is a deubiquitylating enzyme that regulates base excision repair by controlling steady-state levels of DNA polymerase b. Mol. Cell 2011, 41, 609–615. [DOI] [PubMed] [Google Scholar]
- Peschiaroli A.; Skaar J. R.; Pagano M.; Melino G. The ubiquitin specific protease USP47 is a novel beta-TrCP interactor regulating cell survival. Oncogene 2010, 29, 1384–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colland F.; Formstecher E.; Jacq X.; Reverdy C.; Planquette C.; et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 2009, 8, 2286–2295. [DOI] [PubMed] [Google Scholar]
- Guedat P.; Boissy G.; Borg-Capra C.; Colland F.; Daviet L.. et al. Novel cysteine protease inhibitors and their therapeutic applications; Hybrigenics SA: US20070032499, 2007.
- Nicholson B.; Leach C. A.; Goldenberg S. J.; Francis D. M.; Kodrasov M. P.; et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008, 17, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolman H.; Kuipers J.. New thio compounds having fungicidal activity. European Patent Application, 1987, 0234622, A1.
- Basha A.; Lipton M.; Weinreb S. M. A mild, general method for conversion of esters to amides. Tetrahedron Lett. 1977, 18, 4171–4174. [Google Scholar]
- Altun M.; Kramer H. B.; Willems L. I.; McDermott J. L.; Leach C. A.; et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011, 18, 1401–1412. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Lin J.; Su Z. Z.; Collart F. R.; Huberman E.; et al. Induction of differentiation in human promyelocytic HL-60 leukemia cells activates p21, WAF1/CIP1, expression in the absence of p53. Oncogene 1994, 9, 3397–3406. [PubMed] [Google Scholar]
- Ponzio G.; Loubat A.; Rochet N.; Turchi L.; Rezzonico R.; et al. Early G1 growth arrest of hybridoma B cells by DMSO involves cyclin D2 inhibition and p21[CIP1] induction. Oncogene 1998, 17, 1159–1166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.