

Abstract
The clinical response to the antiplatelet prodrug clopidogrel is associated with high intersubject variability and a certain level of therapeutic resistance. Previous studies have suggested that genetic polymorphism of CYP2C19 might be one determinant of clopidogrel efficacy and led to the CYP2C19 genotype-tailored antithrombotic therapy. However, evidence against the role of CYP2C19 from multiple studies implied the involvement of other factors. Here, we report that prodrug activation of the thiophene motif in clopidogrel is attenuated by heavy metabolic attrition of the piperidine motif. CYP3A4/5 was identified to be the enzyme metabolizing the piperidine motif. Inhibiting CYP3A4/5-mediated attrition was shown to potentiate active metabolite formation, which was found to be catalyzed by multiple CYP enzymes. Identifying the significant involvement of CYP3A4/5 and characterizing its mechanistic role in clopidogrel bioactivation might assist future pharmacogenomic studies in exploring the full mechanism underlying clopidogrel efficacy.
Keywords: Clopidogrel resistance, prodrug attrition, CYP3A4/5, active metabolite potentiation, piperidine metabolism
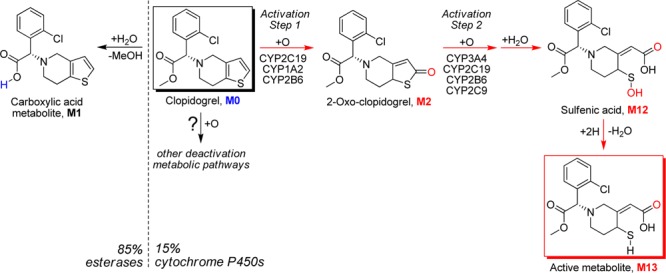
The antiplatelet prodrug clopidogrel is bioactivated by hepatic cytochrome P450 (CYP) enzymes (Scheme 1). The active metabolite binds with a subtype of adenosine diphosphate (ADP) receptor P2Y12 and inhibits ADP-induced platelet aggregation. Clinical antithrombotic responses to clopidogrel are associated with high intersubject variabilities, with 20–40% of patients being classified as nonresponders, poor responders, or resistant to clopidogrel.1−5 Recently, research aiming to rationalize the biological mechanisms underlying the observed clinical uncertainties of clopidogrel therapy has been increasing. Previous in vitro studies showed that CYP2C19 is one of only three CYP enzymes capable of catalyzing active metabolite formation.6 Additionally, clinical research has reported correlation between the CYP2C19 low metabolizer genotype and the decreased antiplatelet response.7,8 On the basis of these findings, CYP2C19 genetic polymorphism is considered to be a key determinant of clopidogrel efficacy.9
Scheme 1. Reported Metabolic Activation and Deactivation Pathways of Clopidogrel.
At the same time, there is also a large body of evidence that does not support the role of CYP2C19 and the CYP2C19-tailored clopidogrel therapy.7,10−13 Ample research results have been reported to support the existence of unidentified determinants, including genetic factors, for clopidogrel efficacy.7,14−16 Recently proposed involvement of paraoxonase-1 in clopidogrel bioactivation17 was debated and ruled out.18−21 To reconcile the current controversy of clinical variability and guide personalized antithrombotic prodrug therapy, characterization of the genetic factors deciding clopidogrel efficacy is highly desired.
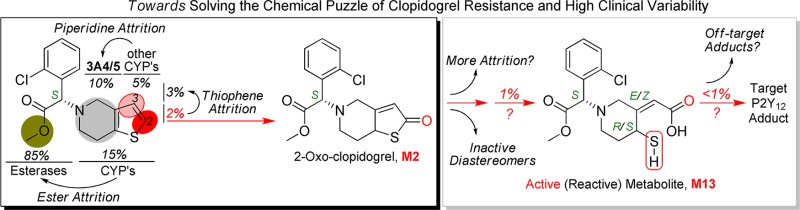
Although the prodrug bioactivation pathway (Scheme 1), conversion from clopidogrel (M0) to 2-oxo-clopidogrel (M2) and to the active thiol metabolite (M13), has been extensively investigated,6,22−24 the complete biotransformation of clopidogrel is devoid of full characterization. The methyl ester of clopidogrel undergoes esterase-catalyzed hydrolysis in the intestine and liver to form the inactive carboxylic acid metabolite (M1), which is the major known route of clopidogrel deactivation and accounts for about 85% of the prodrug loss.25 It has been hypothesized that the remaining clopidogrel goes to hepatic enzymes, mainly CYPs, for the desired metabolic activation. However, the high variability in active metabolite plasma exposures and clinical responses suggests that the active metabolite formation might be accompanied by heavy attrition involving highly variable metabolizing enzymes. To test this hypothesis, we first conducted in vitro metabolic studies of clopidogrel in pooled human liver microsomes (HLM).
In HLM incubation without the CYP cofactor β-nicotinamide adenine dinucleotide 2′-phosphate (reduced, NADPH), a significant portion of clopidogrel (M0) undergoes methyl ester hydrolysis. Because the aim of this study is to characterize the potential nonhydrolyzing deactivation pathways, the selective esterase inhibitor, potassium fluoride, was included in the incubation.26 The extracted ion chromatograms of HLM incubation samples are shown in Figure 1. M2 (+O, M + H+ = 338) possesses an identical LC retention time and MS/MS spectrum to the reference compound 2-oxo-clopidogrel. The structure of M2 was confirmed to be 2-oxo-clopidogrel by an online H-D exchange experiment in which it undergoes just one H-D exchange (Table 1). In addition to M2 formation, metabolite profiling identified eight other NADPH-dependent metabolites, all showing the 35Cl/37Cl isotopic pattern and parent-related fragmentation patterns (Table 1). Two isomeric metabolites of M2 were detected: M3 and M4. Three dehydrogenation metabolites were detected: M5, M6, and M7. M8 showed a mass shift of “+18 Da”, which was confirmed to be “+2H + O” by high-resolution MS analysis. M9 and M10 were proposed to be demethylated products of M5 and M7, respectively.
Figure 1.
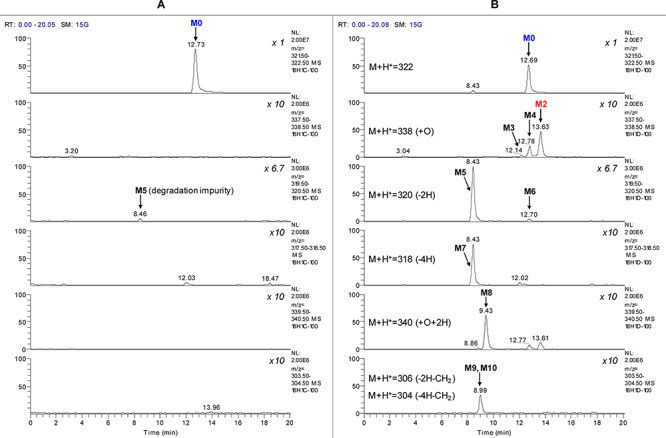
Extracted ion chromatogram of clopidogrel incubation with HLM. (A) Sample without NADPH and (B) sample with NADPH. Nonlabeled peaks are extracted isotopic signals of other metabolite or background signals.
Table 1. Metabolite Profiles of Fragmentation Analysis, Online H-D Exchange, and Deuterated Analogue Experiment Results.
| fragmentation |
D lossb |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| m/z | Δm/z | biotransformation | no. of H-Da | A | B | C | 2-d | 3-d | fragment of metabolism | |
| M0 | 322 | \ | \ | 1 | + | + | \ | |||
| M2 | 338 | +16 | +O | 1 | + | + | \ | y | y | thiophene |
| M3 | 338 | +16 | +O | 2 | + | + | + | n | n | piperidine |
| M4 | 338 | +16 | +O | 2 | + | + | + | n | n | piperidine |
| M5 | 320 | –2 | –2H | 0 | \ | + | + | n | n | piperidine |
| M6 | 320 | –2 | –2H | 1 | \ | + | + | ND | ND | piperidine |
| M7 | 318 | –4 | –4H | 0 | \ | + | \ | n | n | piperidine |
| M8 | 340 | +18 | +O + 2H | 2 | + | + | \ | n | n | thiophene |
| M9 | 306 | –16 | –2H–CH2 | 1 | \ | + | + | n | ND | piperidine |
| M10 | 304 | –18 | –4H–CH2 | 1 | \ | + | + | n | ND | piperidine |
Number of H-D exchange of the charged molecular ion.
Metabolite of the deuterated clopidogrel shows loss of deuterium; ND, metabolite was not detected; fragmentation patterns are illustrated in Figure 2.
MS/MS-based fragmentation analyses and online H-D exchange experiments were conducted to aid the structural elucidation as illustrated in Figure 2. The metabolite profiles were summarized in Table 1. To further pinpoint the location of metabolism (thiophene or piperidine), the 2- and 3-positions of the thiophene ring were labeled with deuterium, and the metabolite profiles of the deuterated clopidogrels were examined for deuterium loss or retention. On the basis of the results of the above analyses, the metabolite structures were tentatively proposed (structural elucidation is further discussed in the Supporting Information). As shown in Scheme 2, we propose that the piperidine ring of clopidogrel undergoes extensive dehydrogenation or hydroxylation to form M3–M7. Similar dehydrogenation and hydroxylation metabolites have been reported for ticlopidine, a close analogue of clopidogrel.27,28 The proposed piperidine hydroxylation metabolite has also been reported as a synthetic intermediate of clopidogrel.29 Metabolism of the thiophene moiety of clopidogrel leads to not only the bioactivation metabolite M2 but also M8, which contains a 3- or 2-hydroxy-2,3-dihydrothiophene substructure. As the result of metabolic attrition, formation of the bioactivation metabolite M2 accounts for only 13% of the CYP-catalyzed conversion.
Figure 2.
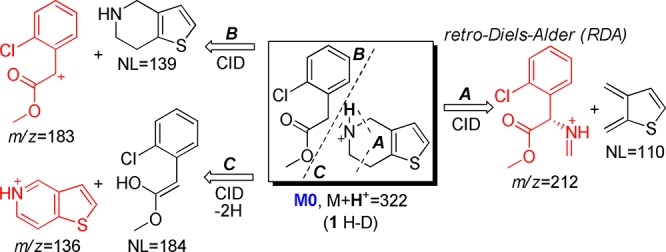
Fragmentation analysis and online H-D exchange experiment result of M0.
Scheme 2. Proposed CYP-Catalyzed Metabolic Pathways of Clopidogrel.
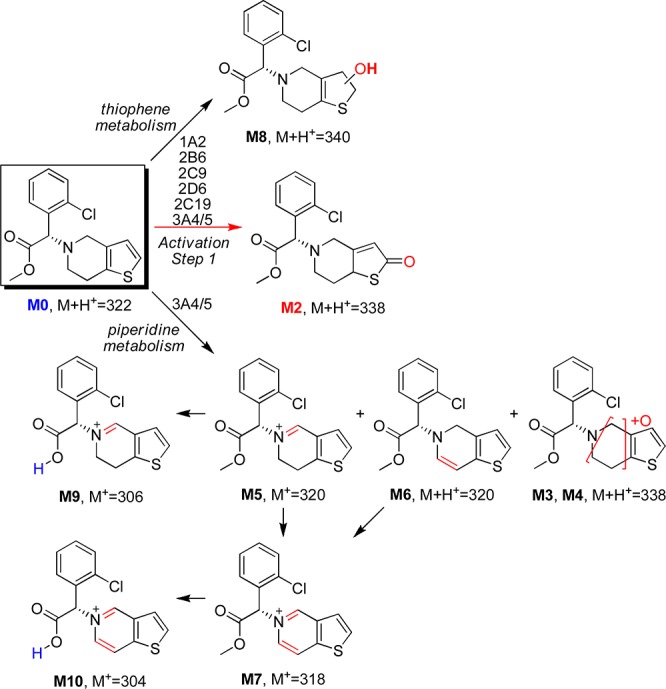
Metabolism studies were then conducted in cDNA-expressed CYP supersomes to phenotype the enzymes that catalyze the metabolite formation. CYP3A4/5 was identified to be the major enzyme catalyzing the piperidine oxidation, while all of the tested CYP enzymes catalyze the thiophene oxidation forming both M2 and M8 (Figure 3 and Figure S3 in the Supporting Information). It has long been believed that the first step of the metabolic activation of clopidogrel, thiophene 2-oxidation leading to M2 formation, is only catalyzed by CYP2C19, CYP1A2, or CYP2B6, as concluded from previous in vitro phenotyping studies in cDNA-expressed CYP supersomes.6 However, the reported experiment contained multiple potential sources that might cause false negative results showing only contributions from the three CYP enzymes. The high CYP isozyme compatibility with clopidogrel revealed here is consistent with its relatively small size and high lipophilicity. Among all of the CYP isozymes, CYP3A4/5 has the most versatile active site to accommodate different substrate binding conformations,30 which explains why piperidine oxidation is only found with CYP3A4/5. It is also important to note that significant attritional metabolism of the thiophene ring is found to accompany 2-oxidation, which further attenuates the desired bioactivation pathway. With more CYP enzymes having been identified to catalyze the formation of M2, the contribution from each individual CYP enzyme is likely to be less significant.
Figure 3.
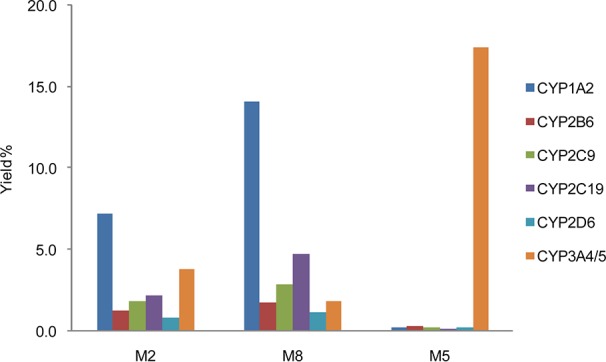
CYP phenotyping results of M2 (metabolic activation), M8 (thiophene metabolism), and M5 (piperidine metabolism) formation.
CYP3A4/5 has been considered as only a negligible player in clopidogrel bioactivation since its direct contribution to the active metabolite formation was found to be insignificant as compared to other CYP enzymes.12,31,32 However, the more detailed characterization of clopidogrel biotransformation uncovers the significant involvement of CYP3A4/5 in affecting the active metabolite formation. As compared to the piperidine oxidation, the thiophene oxidation catalyzed by CYP3A4/5 is minor. Therefore, the major role of CYP3A4/5 is catalyzing prodrug attrition rather than prodrug activation. With a major portion of the absorbed clopidogrel undergoing esterase-catalyzed hydrolysis, the remaining prodrug is partitioned between two groups of CYP enzymes: CYP3A4/5 catalyzes the deactivating piperidine oxidation, while the other CYP enzymes catalyze the thiophene metabolism including the desired 2-oxidation. The partition ratio between the two enzyme groups will decide the active metabolite conversion efficiency and could directly impact the therapeutic effect of clopidogrel. CYP3A4/5 is the most abundant P450 enzyme in human liver. It catalyzes the metabolism of about 50% of drugs used in human therapy and is known to have genetic polymorphism.33,34 Its overall activity can also be highly variable based on drug–drug interaction, food factors, age, etc.35,36 To confirm that the change in CYP3A4/5 activity can impact the clopidogrel bioactivation pathway, we assayed 2-oxo-clopidogrel (M2) formation in HLM with the selective CYP3A4/5 inhibitor ketoconazole (KET). In HLM with KET, the formation of M5 (the major piperidine oxidation metabolite) was found to decrease by 80% as compared to the control sample, which indicates a suppression of the CYP3A4/5-mediated piperidine oxidation pathways. On the other hand, the formation of activation metabolite 2-oxo-clopidogrel (M2) was found to increase by 70%, and M8 formation also increased by 70%, which confirms that the thiophene oxidation is significantly potentiated (Figure 4).
Figure 4.
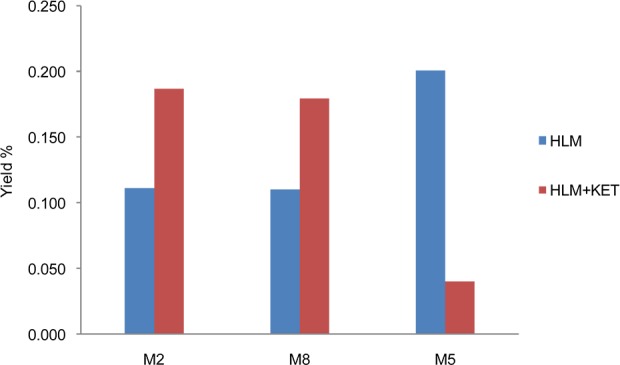
Selective inhibition of CYP3A4/5-catalyzed piperidine metabolism potentiates thiophene bioactivation.
The new mechanistic insight on CYP3A4/5 in clopidogrel bioactivation suggests that an alternative therapeutic strategy to circumvent clopidogrel resistance can be formulated. We propose here that the coadministration of clopidogrel with a selective CYP3A4/5 inhibitor or substrate should be able to divert more clopidogrel from the attrition pathways to the activation pathway, which might lead to the same effect as the loading dose increases and potentiates the active metabolite plasma exposure. An analogous regimen is the anti-HIV therapy of Kaletra (lopinavir/ritonavir),37 which has been used for a decade. In fact, patients might have already benefited from this strategy by taking one alternative therapy recommended by FDA: the concurrent use of clopidogrel with cilostazol, another antiplatelet drug that inhibits type 3 phosphodiesterase.38 Because cilostazol is mainly metabolized by CYP3A4,39 its adjunctive use might decrease the CYP3A4-catalyzed clopidogrel attrition and divert more prodrug to the activation pathway. This hypothesis is supported by recent clinical research results showing that adding cilostazol to clopidogrel therapy improves platelet inhibition in patients with chronic kidney disease (CKD).40 CKD is a known factor of poor clopidogrel responsiveness; the clopidogrel resistance (expressed as P2Y12 inhibition) was significantly alleviated following the concurrent treatment of clopidogrel (75 mg/day) with cilostazol (200 mg/day), while the clopidogrel doubling treatment (150 mg/day) without cilostazol did not show improvement.
The active metabolite (M13) formation from clopidogrel (M0) has been traditionally referred to as “a two-step process” (Scheme 1), which might be an understatement of its mechanistic complexity. Several aspects of this process remain unknown, and the potential involvement of regulatory factors could impact the already-thin active metabolite formation and its plasma exposure. To better understand the biological mechanism underlying clopidogrel efficacy, the following mechanistic details remain to be further investigated (Figure 5): (1) Does the partition between the thiophene metabolism pathways involve reductase? (2) Is the hydrolysis of the thiolactone S-oxide intermediate (M11) in competition with other nucleophiles like glutathione leading to further attrition? (3) Does the active metabolite undergo isomerase-catalyzed tautomerization to its inactive diastereomers? (4) Is the disposition of the active metabolite regulated by transporters? (5) In addition to forming a covalent bond with P2Y12 for the desired efficacy, does the active metabolite form adducts with off-target cellular and plasma proteins leading to diminished half-life or plasma exposure? (6) The active metabolite can be scavenged by glutathione through disulfide bond formation; is the equilibrium dependent on individual oxidative stress status? One or more of the above factors might correlate with certain genotype or disease status and lead to elusive clinical outcomes. The low bioactivation efficiency due to high prodrug attrition might exemplify the impact of the factors on active metabolite plasma exposure.
Figure 5.

Proposed pathways of clopidogrel active metabolite formation with multiple factors that could significantly impact prodrug efficacy.
In summary, we studied the previously unknown metabolic pathways of clopidogrel. Identification of the significant involvement and mechanistic role of CYP3A4/5 in active metabolite formation might help to formulate new therapeutic strategies to battle clopidogrel resistance. Our research results also suggest that factors that impact the partition ratio between the piperidine and the thiophene metabolic pathways might be determinants of clopidogrel efficacy. The more detailed characterization of clopidogrel biotransformation presented here might also facilitate future clinical exploration of using the piperidine and thiophene metabolites as biomarkers to ascertain an individual's status and delineate multicomponent effects on clopidogrel efficacy.
Acknowledgments
We are grateful to Dr. Richard B. Silverman (Northwestern University) for critical reading of the manuscript and Dr. Hongyu Zhao (Abbott Laboratories) for careful revision of the manuscript. We thank Dr. Hua Li (Institute of Pharmacology and Toxicology, Academy of Military Medical Sciences, China) for providing help with the supersome experiment and Xiu Zhang (Analytic Instrumentation Center, Peking University) for helping with NMR analysis. We also thank Pam Beck (Northwestern University) for her assistance.
Glossary
Abbreviations
- CYP
cytochrome P450
- HLM
human liver microsomes
- NADPH
β-nicotinamide adenine dinucleotide 2′-phosphate (reduced)
- KET
ketoconazole
- CKD
chronic kidney disease
Supporting Information Available
Experimental procedures, detailed results, supplementary discussion, supporting schemes and figures, and MS and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Y.Z. made the initial discovery, designed the project, conducted experiments, analyzed the results, and wrote the manuscript. J.Z. conducted the LC-HRMS experiment, assisted with online H-D exchange LC-MS experiments, and contributed to discussion on writing the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Gurbel P. A.; Bliden K. P.; Hiatt B. L.; O'Connor C. M. Clopidogrel for coronary stenting: Response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation 2003, 107, 2908–2913. [DOI] [PubMed] [Google Scholar]
- Michelson A. D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat. Rev. Drug Discovery 2010, 9, 154–169. [DOI] [PubMed] [Google Scholar]
- Kolandaivelu K.; Bhatt D. L. Overcoming resistance to antiplatelet therapy: Targeting the issue of nonadherence. Nat. Rev. Cardiol. 2010, 7, 461–467. [DOI] [PubMed] [Google Scholar]
- Paikin J. S.; Eikelboom J. W.; Cairns J. A.; Hirsh J. New antithrombotic agents-insights from clinical trials. Nat. Rev. Cardiol. 2010, 7, 498–509. [DOI] [PubMed] [Google Scholar]
- Angiolilloa D. J.; Fernandez-Ortizb A.; Bernardob E.; Ramírezb C.; Barrera-Ramirezb C.; Sabatéb M.; Hernándezb R.; Morenob R.; Escanedb J.; Alfonsob F.; Bañuelosb C.; Costaa M. A.; Bassa T. A.; Macaya C. Identification of low responders to a 300-mg clopidogrel loading dose in patients undergoing coronary stenting. Thromb. Res. 2005, 115, 101–108. [DOI] [PubMed] [Google Scholar]
- Kazui M.; Nishiya Y.; Ishizuka T.; Hagihara K.; Farid N. A.; Okazaki O.; Ikeda T.; Kurihara A. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab. Dispos. 2010, 38, 92–99. [DOI] [PubMed] [Google Scholar]
- Shuldiner A. R.; O'Connell J. R.; Bliden K. P.; Gandhi A.; Ryan K.; Horenstein R. B.; Damcott C. M.; Pakyz R.; Tantry U. S.; Gibson Q.; Pollin T. I.; Post W.; Parsa A.; Mitchell B. D.; Faraday N.; Herzog W.; Gurbel P. A. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. J. Am. Med. Assoc. 2009, 302, 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt J. T.; Close S. L.; Iturria S. J.; Payne C. D.; Farid N. A.; Ernest C. S. 2nd; Lachno D. R.; Salazar D.; Winters K. J. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J. Thromb. Haemostasis 2007, 5, 2429–2436. [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration. FDA Drug Safety Communication: Reduced effectiveness of Plavix (clopidogrel) in patients who are poor metabolizers of the drug. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm203888.htm, 2010.
- Paré G.; Mehta S. R.; Yusuf S; Anand S. S.; Connolly S. J.; Hirsh J.; Simonsen K.; Bhatt D. L.; Fox K. A. A.; Eikelboom J. W. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N. Engl. J. Med. 2010, 363, 1704–1714. [DOI] [PubMed] [Google Scholar]
- Bauer T.; Bouman H. J.; van Werkum J. W.; Ford N. F.; ten Berg J. M.; Taubert D. Impact of CYP2C19 variant genotypes on clinical efficacy of antiplatelet treatment with clopidogrel: Systematic review and meta-analysis. Br. Med. J. 2011, 343, d4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasar U.; Bennet A. M.; Eliasson E.; Lundgren S.; Wiman B.; De Faire U.; Rane A. Allelic variants of cytochromes P450 2C modify the risk for acute myocardial infarction. Pharmacogenetics 2003, 13, 715–720. [DOI] [PubMed] [Google Scholar]
- Barker C. M.; Murray S. S.; Teirstein P. S.; Kandzari D. E.; Topol E. J.; Price M. J. Pilot study of the antiplatelet effect of increased clopidogrel maintenance dosing and its relationship to CYP2C19 genotype in patients with high on-treatment reactivity. J. Am. Coll. Cardiol. Interv. 2010, 3, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Hulot J. S.; Collet J. P.; Silvain J.; Pena A.; Bellemain-Appaix A.; Barthélémy O.; Cayla G.; Beygui F.; Montalescot G. Cardiovascular risk in clopidogrel-treated patients according to cytochrome P450 2C19*2 loss-of-function allele or proton pump inhibitor coadministration: a systematic meta-analysis. J. Am. Coll. Cardiol. 2010, 56, 134–143. [DOI] [PubMed] [Google Scholar]
- Holmes D. R. Jr.; Dehmer G. J.; Kaul S.; Leifer D.; O'Gara P. T.; Stein C. M. ACCF/AHA Clopidogrel clinical alert: Approaches to the FDA boxed warning”: A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010, 122, 537–557. [DOI] [PubMed] [Google Scholar]
- Fontana P.; Berdagué P.; Castelli C.; Nolli S.; Barazer I.; Fabbro-Peray P.; Schved J. F.; Bounameaux H.; Mach F.; de Moerloose P.; Reny J. L. Clinical predictors of dual aspirin and clopidogrel poor responsiveness in stable cardiovascular patients from the ADRIE study. J. Thromb. Haemostasis 2010, 8, 2614–2623. [DOI] [PubMed] [Google Scholar]
- Bouman H. J.; Schömig E.; van Werkum J. W.; Velder J.; Hackeng C. M.; Hirschhäuser C.; Waldmann C.; Schmalz H. G.; ten Berg J. M.; Taubert D. Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat. Med. 2011, 17, 110–116. [DOI] [PubMed] [Google Scholar]
- Cuisset T.; Morange P. E.; Quilici J.; Bonnet J. L.; Gachet C.; Alessi M. C. Paraoxonase-1 and clopidogrel efficacy. Nat. Med. 2011, 17, 1039. [DOI] [PubMed] [Google Scholar]
- Dansette P. M.; Rosi J.; Bertho G.; Mansuy D. Paraoxonase-1 and clopidogrel efficacy. Nat. Med. 2011, 17, 1040–1041. [DOI] [PubMed] [Google Scholar]
- Dansette P. M.; Rosi J.; Bertho G.; Mansuy D. Cytochromes P450 Catalyze Both Steps of the Major Pathway of Clopidogrel Bioactivation, whereas Paraoxonase Catalyzes the Formation of a Minor Thiol Metabolite Isomer. Chem. Res. Toxicol. 2012, 25, 348–356. [DOI] [PubMed] [Google Scholar]
- Gong I. Y.; Crown N.; Suen C. M.; Schwarz U. I.; Dresser G. K.; Knauer M. J.; Sugiyama D.; Degorter M. K.; Woolsey S.; Tirona R. G.; Kim R. B.. Clarifying the importance of CYP2C19 and PON1 in the mechanism of clopidogrel bioactivation and in vivo antiplatelet response. Eur. Heart. J. Published online February 27, 2012; DOI: 10.1093/eurheartj/ehs042. [DOI] [PubMed] [Google Scholar]
- Dansette P. M.; Libraire J.; Bertho G.; Mansuy D. Metabolic oxidative cleavage of thioesters: evidence for the formation of sulfenic acid intermediates in the bioactivation of the antithrombotic prodrugs ticlopidine and clopidogrel. Chem. Res. Toxicol. 2009, 22, 369–373. [DOI] [PubMed] [Google Scholar]
- Dansette P. M.; Thebault S.; Bertho G.; Mansuy D. Formation and fate of a sulfenic acid intermediate in the metabolic activation of the antithrombotic prodrug prasugrel. Chem. Res. Toxicol. 2010, 23, 1268–1274. [DOI] [PubMed] [Google Scholar]
- Shan J.; Zhang B.; Zhu Y.; Jiao B.; Zheng W.; Qi X.; Gong Y.; Yuan Y.; Lv F.; Sun H. Overcoming clopidogrel resistance: discovery of vicagrel as a highly potent and orally bioavailable antiplatelet agent. J. Med. Chem. 2012, 55, 3342–3352. [DOI] [PubMed] [Google Scholar]
- Caplain H.; Donat F.; Gaud C.; Necciari J. Pharmacokinetics of clopidogrel. Semin. Thromb. Hemostasis 1999, 25Suppl. 225–28. [PubMed] [Google Scholar]
- Pereillo J.; Maftouh M.; Andrieu A.; Uzabiaga M.; Fedeli O.; Savi P.; Pascal M.; Herbert J.; Maffrand J.; Picard C. Structure and stereochemistry of the active metabolite of clopidogrel. Drug Metab. Dispos. 2002, 30, 1288–1295. [DOI] [PubMed] [Google Scholar]
- Dalvie D. K.; O'Connell T. N. Characterization of novel dihydrothienopyridinium and thienopyridinium metabolites of ticlopidine in vitro: Role of peroxidases, cytochromes p450, and monoamine oxidases. Drug Metab. Dispos. 2004, 32, 49–57. [DOI] [PubMed] [Google Scholar]
- Picard-Fraire C. Pharmacokinetic and metabolic characteristics of ticlopidine in relation to its inhibitory properties on platelet function. Agents Actions Suppl. 1984, 15, 68–75. [PubMed] [Google Scholar]
- Horne S. E.; Weeratunga G.; Comanita B. M.; Nagireddy J. R.; McConachie L. K.. Process for the preparation of tetrahydrothieno[3,2-c]pyridine derivatives. U.S. Patent 6,495,691 B1, December 17, 2002.
- Ohkura K.; Kawaguchi Y.; Watanabe Y.; Masubuchi Y.; Shinohara Y.; Hori H. Flexible structure of cytochrome P450: Promiscuity of ligand binding in the CYP3A4 heme pocket. Anticancer Res. 2009, 29, 935–942. [PubMed] [Google Scholar]
- Giusti B.; Gori A. M.; Marcucci R.; Saracini C.; Sestini I.; Paniccia R.; Valente S.; Antoniucci D.; Abbate R.; Gensini G. F. Cytochrome P450 2C19 loss-of-function polymorphism, but not CYP3A4 IVS10 + 12G/A and P2Y12 T744C polymorphisms, is associated with response variability to dual antiplatelet treatment in high-risk vascular patients. Pharmacogenet. Genomics 2007, 17, 1057–1064. [DOI] [PubMed] [Google Scholar]
- Clarke T. A.; Waskell L. A. The metabolism of clopidogrel is catalyzed by human cytochrome P450 3A and is inhibited by atorvastatin. Drug Metab. Dispos. 2003, 31, 53–59. [DOI] [PubMed] [Google Scholar]
- Lee S. J.; Goldstein J. A. Functionally defective or altered CYP3A4 and CYP3A5 single nucleotide polymorphisms and their detection with genotyping tests. Pharmacogenomics 2005, 6, 357–371. [DOI] [PubMed] [Google Scholar]
- Hustert E.; Haberl M.; Burk O.; Wolbold R.; He Y. Q.; Klein K.; Nuessler A. C.; Neuhaus P.; Klattig J.; Eiselt R.; Koch I.; Zibat A.; Brockmöller J.; Halpert J. R.; Zanger U. M.; Wojnowski L. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics 2001, 11, 773–779. [DOI] [PubMed] [Google Scholar]
- Lamba J. K.; Lin Y. S.; Schuetz E. G.; Thummel K. E. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Delivery Rev. 2002, 54, 1271–1294. [DOI] [PubMed] [Google Scholar]
- Dorne J. L.; Walton K.; Renwick A. G. Human variability in CYP3A4 metabolism and CYP3A4-related uncertainty factors for risk assessment. Food Chem. Toxicol. 2003, 41, 201–224. [DOI] [PubMed] [Google Scholar]
- Sham H. L.; Kempf D. J.; Molla A.; Marsh K. C.; Kumar G. N.; Chen C. M.; Kati W.; Stewart K.; Lal R.; Hsu A.; Betebenner D.; Korneyeva M.; Vasavanonda S.; McDonald E.; Saldivar A.; Wideburg N.; Chen X.; Niu P.; Park C.; Jayanti V.; Grabowski B.; Granneman G. R.; Sun E.; Japour A. J.; Leonard J. M.; Plattner J. J.; Norbeck D. W. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob. Agents Chemother. 1998, 42, 3218–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y.; Tani T.; Kanbe T.; Watanabe K. Effects of cilostazol on platelet aggregation and experimental thrombosis. Arzneimittelforschung 1985, 35, 1144–1149. [PubMed] [Google Scholar]
- Abbas R.; Chow C. P.; Browder N. J.; Thacker D.; Bramer S. L.; Fu C. J.; Forbes W.; Odomi M.; Flockhart D. A. In vitro metabolism and interaction of cilostazol with human hepatic cytochrome P450 isoforms. Hum. Exp. Toxicol. 2000, 19, 178–184. [DOI] [PubMed] [Google Scholar]
- Woo J. S.; Kim W.; Lee S. R.; Jung K. H.; Kim W. S.; Lew J. H.; Lee T. W.; Lim C. K. Platelet reactivity in patients with chronic kidney disease receiving adjunctive cilostazol compared with a high-maintenance dose of clopidogrel: Results of the effect of platelet inhibition according to clopidogrel dose in patients with chronic kidney disease (PIANO-2 CKD) randomized study. Am. Heart J. 2011, 162, 1018–1025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.