

Abstract

Neurotrophins and their receptors (TRKs) play key roles in the development of the nervous system and the maintenance of the neural network. Accumulating evidence points to their role in malignant transformations, chemotaxis, metastasis, and survival signaling and may contribute to the pathogenesis of a variety of tumors of both neural and non-neural origin. By screening the GNF kinase collection, a series of novel oxindole inhibitors of TRKs were identified. Optimization led to the identification of GNF-5837 (22), a potent, selective, and orally bioavailable pan-TRK inhibitor that inhibited tumor growth in a mouse xenograft model derived from RIE cells expressing both TRKA and NGF. The properties of 22 make it a good tool for the elucidation of TRK biology in cancer and other nononcology indications.
Keywords: neurotrophins, NGF, tropomyosin receptor kinase, TRK, oxindole, GNF-5837
The tropomyosin receptor kinase (TRK) family includes three members of highly homologous receptor tyrosine kinases, TRKA, TRKB, and TRKC (also known as NTRK1, NTRK2, and NTRK3), that specifically bind to neurotrophins. The binding of neutrophins to the extracellular domains of TRK causes the kinase to autophosphorylate at several intracellular tyrosine sites and triggers downstream signals involving RAS/ERK, PI3K/AKT, and PLC-γ1 transduction pathways. The TRK receptors and their respective ligands are implicated in proliferation, survival, differentiation, and functional regulation of different neuronal populations in the nervous system, especially during embryogenesis.1 In addition to their physiological role in neuronal and non-neuronal tissues, the neurotrophin receptors are found to be involved in the development and progression of cancer.2−9 Several different mechanisms have been proposed and described, which involve oncogenic activation of TRK receptors, including over/ectopic expression and stimulation by autocrine/paracrine loops, expression of alternative and constitutively active splice variants under hypoxia conditions, point mutations, and genomic rearrangements.2 A TRKA rearrangement leading to a constitutively active and ligand-independent fused protein TPM3-TRKA was originally identified in a colorectal carcinoma biopsy11−13 and later found to be associated to a subset of papillary thyroid carcinomas (PTC).14−16 Also, the TRKC fusion protein Tel-TRKC has been identified as the causative event in rare cancers such as secretory breast carcinoma (SBC),17 congenital fibrosarcoma (CFS),18 and congenital mesoblastic nefroma (CMN).19 In addition, there may be a role for small molecule inhibitors in other cancer settings; for example, TRKB has been reported as a strong predictor of aggressive tumor growth in neuroblastoma, and it is also associated with increase resistance to chemotherapy and anoikis.20,21 Moreover, high and increased expression of TRK receptors in pancreatic cancer correlates to tumor progression, perineural invasion, pain, and metastasis.7,22−24
Several very potent nonselective TRK inhibitors have been described, and some of them have entered the clinic.25 To date, only Array BioPharma has disclosed data associated with selective pan-TRK inhibitors, but their exact structures have not been published.26 To investigate the role of TRKs in cancer biology, we set out to identify selective inhibitors with appropriate pharmacokinetic profile to be studied in in vitro and in vivo systems.
The GNF kinase compound collection was screened for antiproliferative effects in a Ba/F3 assay where Ba/F3 cells were rendered TRKC dependent and IL-3 independent by overexpressing the constitutively active Tel-TRKC fusion. The screen identified oxindole 4 (Scheme 1) as a TRKC inhibitor as it showed differential cytotoxicity as compared to parental Ba/F3 cells grown in the presence of IL-3. Compound 4, originally synthesized by morphing sunitinib with other kinase inhibitors to further occupy the hydrophobic pocket,27 was shown to inhibit Ba/F3-Tel-TRKA, TRKB, and TRKC with an IC50 less than 0.06 μM (Table 1). The cellular kinase selectivity profile of 4 was determined in a Ba/F3 panel using Ba/F3 cells rendered IL-3 independent by stable transduction with 25 selected kinases activated by fusion with a dimerizing protein partner (e.g., Bcr or Tel).28 In this panel, compound 4 showed a significant inhibitory activity only for FMS, KDR, c-Kit, and PDGFRβ (Tables 1 and 2).
Scheme 1. Preparation of Oxindole Amides 1–11.

Reagents and conditions: (a) CH2(CO2Me)2, NaH, DMSO, 60 °C, 99%. (b) AcOH, HCl, 110 °C, 99%. (c) EtOH, concentrated H2SO4, reflux, 96%. (d) 3-Nitroaniline, Pd(OAc)2, Xantphos, Cs2CO3, 80 °C, 85%. (e) H2 (balloon), 10% Pd/C, AcOH, room temperature, 72%. (f) R-COOH, HATU, DIEA, DMF, room temperature, 47–99%. (g) 1H-pyrrole-2-carbaldehyde, EtOH, 80 °C, 35–91%.
Table 1. Cellular and Enzymatic Activities of Oxindole Amides and Ureas.
| IC50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| cellular Ba/F3
assaysa |
biochemical assaysc |
|||||||
| TRK |
TRK |
|||||||
| compd | A | B | C | KDR | WTb | A | B | C |
| 1 | 2.3 | 1.3 | 1.2 | 1.0 | 1.3 | NA | NA | 1.9 |
| 2 | 0.91 | 0.68 | 0.69 | 0.60 | 0.93 | 0.16 | NA | NA |
| 3 | 0.85 | 0.66 | 0.57 | NA | 2.2 | NA | NA | NA |
| 4 | 0.059 | 0.037 | 0.023 | 0.50 | 4.8 | 0.007 | NA | 0.029 |
| 5 | 0.015 | 0.005 | 0.007 | 0.50 | >10 | 0.007 | 0.054 | NA |
| 6 | 0.39 | 0.22 | 0.27 | 2.8 | 4.7 | 0.016 | NA | 0.097 |
| 7 | 0.013 | 0.012 | 0.012 | 0.50 | 4.5 | 0.007, | 0.007 | NA |
| 8 | 0.36 | 0.55 | 0.44 | 0.70 | 2.1 | 0.027 | NA | NA |
| 9 | 0.034 | 0.019 | 0.029 | 0.61 | 4.9 | 0.003 | 0.06 | NA |
| 10 | 0.19 | 0.079 | 0.15 | 4.9 | >10 | 0.068 | 0.035 | NA |
| 11 | 0.022 | 0.020 | NA | 0.20 | 4.3 | NA | NA | NA |
| 12 | >10 | >10 | >10 | >10 | >10 | NA | NA | >10 |
| 13 | 0.36 | 0.12 | 0.19 | 2.2 | >10 | 0.049 | NA | NA |
| 14 | 0.014 | 0.005 | 0.008 | 0.60 | 1.3 | NA | 0.027 | NA |
| 15 | >10 | >10 | >10 | >10 | >10 | NA | NA | NA |
| 16 | 0.01 | 0.003 | 0.006 | 1.5 | >10 | NA | 0.017 | NA |
| 17 | 0.009 | 0.006 | 0.022 | 0.23 | 5.2 | 0.004 | 0.019 | NA |
| 18 | 0.012 | 0.004 | 0.004 | 1.3 | 4.6 | 0.009 | NA | NA |
| 19 | 0.013 | 0.011 | 0.027 | NA | 1.4 | NA | 0.015 | NA |
| 20 | 0.004 | 0.005 | 0.004 | 0.52 | 2.4 | 0.007 | NA | 0.04 |
| 21 | 0.05 | 0.017 | 0.018 | 3.3 | 3.3 | 0.009 | NA | NA |
| 22 | 0.011 | 0.009 | 0.007 | 3.0 | 5.6 | 0.008 | 0.012 | NA |
Proliferation assays with Tel-TRK or Tel-KDR fusions.
Proliferation assay using parental Ba/F3 cells.
HTRF or Caliper assays with recombinant TRKA, -B, and -C kinase domains.
Table 2. Ba/F3 Cellular Kinase Panel (IC50, μM).
| Ba/F3a | 4 | 5 | 7 | 16 | 20 | 22 |
|---|---|---|---|---|---|---|
| ABLb | 6.4 | 5.2 | 3.8 | 8.2 | 1.8 | 8.2 |
| ALKc | 6.5 | 5.7 | 3.0 | >10 | 7.1 | 6.8 |
| ALK | 5.9 | 5.3 | NA | 8.8 | 2.0 | 3.4 |
| BMX | 7.2 | 6.7 | 4.9 | >10 | 3.3 | 6.7 |
| EPHA3 | 5.2 | 5.1 | NA | 5.4 | 1.9 | 5.9 |
| EPHB2 | 2.4 | 4.5 | NA | 2.3 | 1.7 | 5.8 |
| FGFR3 | 6.3 | 7.7 | NA | >10 | 1.7 | 9.3 |
| FGFR4 | 5.4 | 7.0 | NA | >10 | 1.9 | 7.0 |
| FGR | >10 | 5.9 | NA | 6.3 | 2.0 | 3.6 |
| FLT1 | 2.2 | >10 | 3.6 | 3.2 | 1.5 | 4.4 |
| FLT3 | 9.4 | 4.7 | 6.4 | 5.5 | 2.1 | 5.1 |
| FMS | 0.31 | 4.2 | 3.7 | 7.6 | 3.4 | 4.0 |
| IGF1R | 5.6 | 6.2 | 5.7 | >10 | 1.5 | 7.6 |
| INSR | 5.8 | 5.7 | 5.4 | >10 | 2.9 | 8.0 |
| JAK2 | 3.1 | 5.8 | 5.1 | 5.6 | 1.9 | 2.9 |
| KIT | 0.021 | 0.013 | 0.5 | 0.046 | 0.13 | 0.91 |
| MET | 9.6 | >10 | 5.1 | NA | 1.5 | 5.8 |
| LYN | 7.1 | 6.6 | 6.0 | >10 | 1.9 | 7.3 |
| PDGFRβ | 0.002 | 0.1 | 0.2 | NA | 0.12 | 0.87 |
| RET | 1.1 | 0.18 | 0.8 | 0.1 | 1.0 | 5.3 |
| RON | 5.8 | >10 | 4.9 | >10 | 2.7 | 4.7 |
| ROS | 5.5 | 5.7 | 6.0 | 7.0 | 1.8 | 6.0 |
| SRC | 5.5 | 5.8 | 5.3 | >10 | 2.8 | 7.3 |
| TIE1 | 7.9 | 5.4 | 5.9 | 7.0 | 2.9 | 5.7 |
| ZAP70 | 4.8 | >10 | 6.0 | NA | 2.3 | 4.7 |
Ba/F3 cells rendered IL-3 independent by stable transduction with the indicated kinase fused with a Tel dimerization partner unless otherwise specified.
Bcr-abl.
NMP-ALK.
Medicinal chemistry optimization was initiated with the goal of identifying a compound with increased selectivity over other kinases, especially KDR, and with appropriate physicochemical and pharmacokinetic properties to support in vivo studies. New compounds were tested for antiproliferative effects in Ba/F3-Tel-TRKA/B/C and Ba/F3-Tel-KDR assays, and the cytotoxicity effects in those assays were compared with effects in parental wild-type Ba/F3 cells (WT). In addition, selected compounds were tested in biochemical assays using either homogeneous time-dependent fluorescence (HTRF) or Caliper method assays with recombinant TRKA, -B, and -C kinase domains (Table 1) to confirm activity.
Compounds 1–22 are described in Schemes 1–3. Investigative structure–activity relationship (SAR) of the amide postulated to be occupying the hydrophobic pocket led to the conclusion that the 3-trifluoromethylphenyl-amide is needed to achieve potent cellular TRK inhibitory activity. The unsubstituted benzamide 2 exhibited only very modest activity and replacement of the trifluoromethyl group with a fluorine atom as in 3 led to a decrease in activity. Compound 1, with an acetamide in lieu of a benzamide, exhibited activity similar to that on wild-type cells, indicating loss of specific inhibitory activity for TRK. In an attempt to improve the solubility of the lead compound, the incorporation of heterocyclic groups on the phenyl ring was assessed (5–11). Several basic groups at the 5-position were well tolerated, preferentially in conjunction with the 3-trifluoromethyl group (compare methyl analogue 6 to trifluoromethyl derivative 7). The piperazine at the 4-position led to a drop in activity when compared with the same substitution in the 5-position (8 vs 7). In all cases, the solubility remained low. The elimination of the carbonyl contact to give aniline compound 12 resulted in complete loss of activity as expected for a classic type II DFG-out inhibitor where the amide makes a hydrogen bond contact with the DFG aspartate and glutamate in helix c.27 Replacement of the pyrrole with other groups (pyrrolidine, isooxazole, and cyclopentane) resulted in loss of activity. Methylation of the pyrrole nitrogen of compound 5 yielded a significantly less potent compound (13), suggesting that the pyrrole NH may be involved in a key interaction at the protein hinge region. Decoration on the pyrrole ring led to compounds of varying potency (14–19). The addition of an amide group on the pyrrole at the 4-position (sunitinib like), with or without additional methyl groups, yielded compounds with good potency but poor selectivity against KDR (17 and 19). Excellent potency and selectivity were obtained with the corresponding 4-carboxylic acids (16 and 18). Nitrile replacement of the 4-carboxylic acid (14) was also tolerated, but selectivity was not optimal. In contrast, substitution on the pyrrole with an acid in the 5-position (15) was not tolerated. Replacement of the amide in compound 4 with a urea functionality led to compounds with increased potency and good selectivity against KDR with or without methyl decoration on the central phenyl ring (20 and 21). The addition of a fluorine in the 2-position of the trifluoromethylphenyl ring (22, GNF-5837) further enhanced selectivity over KDR.
Scheme 3. Preparation of Oxindole Ureas 20–22.

Reagents and conditions: (a) Corresponding isocyanate, TEA, THF, room temperature, 83–94%. (b) 1H-pyrrole-2-carbaldehyde, EtOH, 80 °C, 63–87%.
Scheme 2. Preparation of Oxindole Amides with Substituted Pyrroles 14–19.

Reagents and conditions: (a) 3-(4-Methylpiperazin-1-yl)-5-(trifluoromethyl)benzoic acid, HATU, DIEA, DMF, room temperature, 75%. (b) Corresponding aldehyde, EtOH, 80 °C, 49–80%. (c) Compound 16 to 17 and 18 to 19, corresponding amine, HATU, DIEA, DMF, room temperature, 45–70%.
Compound 20 was cocrystallized with TRKC and was found to bind to the unactivated kinase (DFG-out) (Figure 1).29 The peptide backbone from the hinge residues E618, Y619, and M620 formed key hydrogen bonds to the heterocycles of 20. A second set of polar interactions involved the compound's urea functionality, which was hydrogen bonded to the side chain of E588 from the α-C-helix and to the peptide backbone of D697 from the DFG motif. The interactions of the urea moiety in this costructure were very similar to those documented for other urea derivatives used in kinase inhibitor design.30−33 In addition to these polar interactions, two aromatic–aromatic interactions were recognized involving the compound's aminophenyl ring: a stacking interaction with the phenyl ring from the gatekeeper F617 and a face to edge interaction with F698 from the DFG motif.
Figure 1.
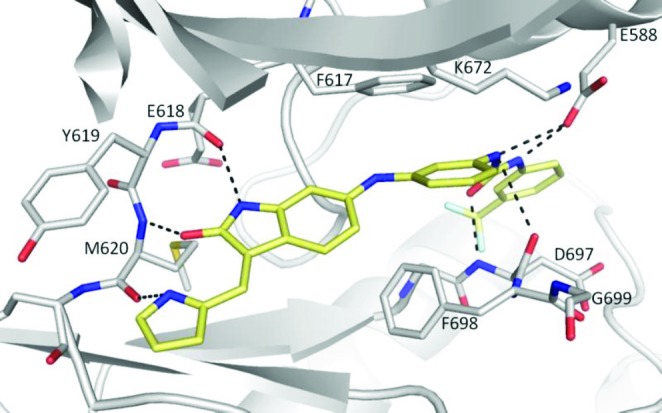
X-ray crystal structure of compound 20 (stick representation; carbon in yellow, oxygen in red, nitrogen in blue, and fluorine in light blue) binding to the active site of TRKC kinase domain. The kinase domain is depicted as a cartoon with carbon atoms in gray, nitrogen in blue, and oxygen in red. Residues of the hinge region, the DFG motif, K672, and E588 are shown as sticks. Hydrogen bonds are shown as dashed lines.
By virtue of their promising selectivity against KDR, compounds 5, 7, 16, 20, and 22 were tested in a Ba/F3 panel containing 25 additional kinases (Table 2). All of the compounds inhibited both c-Kit and PDGFRβ to varying degrees.
The cellular anti-TRKA activity of compound 22 was further determined in Ba/F3 and RIE (rat intestinal epithelial) cells engineered to express both TRKA and NGF. In Ba/F3 cells expressing TRKA and NGF, 22 demonstrated potent antiproliferation activity with IC50 of 0.042 μM, whereas it did not have antiproliferative activity up to 10 μM in parental Ba/F3 cells whose proliferation is dependent on IL-3. RIE cells expressing TRKA and NGF are able to grow and proliferate under low attachment condition and are resistant to detachment-induced apoptosis (anoikis). Compound 22 potently inhibited cell growth and proliferation of RIE cells expressing TRKA and NGF under the low attachment condition with an IC50 of 0.017 μM.
Inhibitory activities for c-Kit and PDGFR were confirmed in cellular assays in Mo7e cells and Rat-A10 cells, respectively. In those assays, compound 22 showed activity in the submicromolar to micromolar range (IC50 Mo7e-c-Kit, 1 μM; IC50 Rat-A10-PDGFR, 0.5 μM), allowing for a significant selectivity window for TRK over c-Kit and PDGFR (IC50RIE-NGF, 0.017 μM).
To determine the in vivo PK data, compound 22 was administered intravenously to male Balb/c mice and Sprague–Dawley rats, and the drug clearance was found to be low and the volume of distribution moderate (mice) to high (rat). When administered orally by gavage, it gave moderate biovailability in both mice and rats due to poor absorption deriving from a combination of poor permeability and low aqueous solubility (Table 3). In the mice, the drug concentration in the brain after oral delivery was below the limit of quantitation, suggesting that the compound does not effectively cross the blood–brain barrier.
Table 3. In Vivo PK Properties of Compound 22.
| species | Balb/c mice | Sprague–Dawley rats | ||
|---|---|---|---|---|
| routea | iv | po | iv | po |
| dose (mg/kg) | 5 | 20 | 3 | 10 |
| AUC (h nM) | 19339 | 14293 | 10616 | 6837 |
| CL (mL/min/kg) | 8.2 | 9 | ||
| Vss (L/kg) | 1.4 | 3.6 | ||
| Cmax (nM) | 16360 | 1173 | 5196 | 449 |
| T1/2 (h) | 4.4 | 4.5 | 7.5 | 7.1 |
| F (%) | 18 | 19 | ||
Formulated in PEG300:D5W/3:1 as a solution.
To demonstrate in vivo efficacy, compound 22 was administered at ascending doses once daily for 10 days in mice (Figure 2) with established tumor xenografts derived from RIE cells expressing both TRKA and NGF. In this study, 72 and 100% tumor regression was observed at 50 and 100 mg/kg, respectively. At 25 mg/kg, only partial tumor growth inhibition was achieved.34
Figure 2.
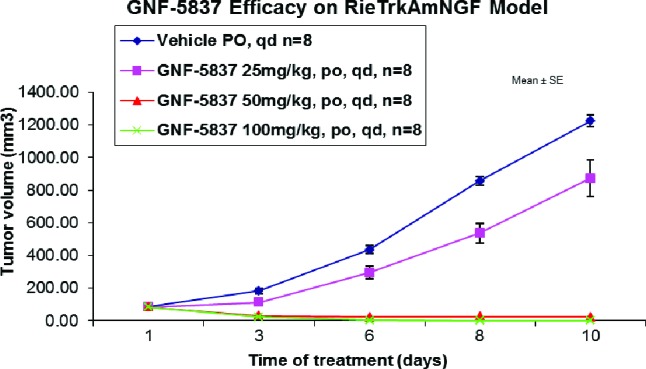
Efficacy of GNF-5837 (22) in Rie-TRKAmNGF xenograft model.
In summary, we have identified a pan TRK inhibitor with excellent selectivity against other kinases, acceptable pharmacokinetic properties, and proven efficacy in vivo. The properties of 22 make it a good tool for the elucidation of TRK biology in cancer and other disease areas. In addition, we have disclosed the first crystal structure of TRKC with a TRKC inhibitor, which has the potential to aid the design of other classes of selective TRK inhibitors.
Acknowledgments
We thank Eric Peters and Daniel Mason for HRMS data and David Jones for NMR data.
Supporting Information Available
Experimental procedures and characterization of compounds 4, 5, 7, 16, 20, and 22; kinase selectivity and ADMET data for compound 22; and details of in vitro and in vivo assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Huang E. J.; Reichardt L. F. TRK receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [DOI] [PubMed] [Google Scholar]
- Pierotti M. A.; Greco A. Oncogenic rearrangements of the NTRK1/NGF receptor. Cancer Lett. 2006, 232, 90–98. [DOI] [PubMed] [Google Scholar]
- Lei L.; Parada L. F. Transcriptional regulation of TRK family neurotrophin receptors. Cell. Mol. Life Sci. 2007, 64, 522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadec C.; Meignan S.; Adriaenssens E.; Foveau B.; Vanhecke E.; Romon R.; Toillon R. A.; Oxombre B.; Hondermarck H.; Le Bourhis X. TRKA overexpression enhances growth and metastasis of breast cancer cells. Oncogene 2009, 28, 1960–1970. [DOI] [PubMed] [Google Scholar]
- Borrello M. G.; Bongarzone I.; Pierotti M. A.; Luksch R.; Gasparini M.; Collini P.; Pilotti S.; Rizzetti M. G.; Mondellini P.; De Bernardi B.; Di Martino D.; Garaventa A.; Brisigotti M.; Tonini G. P. TRK and ret proto-oncogene expression in human neuroblastoma specimens: High frequency of TRK expression in non-advanced stages. Int. J. Cancer 1993, 54, 540–545. [DOI] [PubMed] [Google Scholar]
- Ma J.; Jiang Y.; Jiang Y.; Sun Y.; Zhao X. Expression of nerve growth factor and tyrosine kinase receptor A and correlation with perineural invasion in pancreatic cancer. J. Gastroenterol. Hepatol. 2008, 23, 1852–1859. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Friess H.; diMola F. F.; Zimmermann A.; Graber H. U.; Korc M.; Buchler M. W. Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J. Clin. Oncol. 1999, 17, 2419–2428. [DOI] [PubMed] [Google Scholar]
- Festuccia C.; Muzi P.; Gravina G. L.; Millimaggi D.; Speca S.; Dolo V.; Ricevuto E.; Vicentini C.; Bologna M. Tyrosine kinase inhibitor CEP-701 blocks the NTRK1/NGF receptor and limits the invasive capability of prostate cancer cells in vitro. Int. J. Oncol. 2007, 30, 193–200. [PubMed] [Google Scholar]
- Davidson B.; Reich R.; Lazarovici P.; Nesland J. M.; Skrede M.; Risberg B.; Trope C. G.; Florenes V. A. Expression and activation of the nerve growth factor receptor TRKA in serous ovarian carcinoma. Clin. Cancer Res. 2003, 9, 2248–2259. [PubMed] [Google Scholar]
- Thiele C. J.; Li Z.; McKee A. E. On TRK—The TRKB signal transduction pathway is an increasingly important target in cancer biology. Clin. Cancer Res. 2009, 15, 5962–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Zanca D.; Hughes S. H.; Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 1986, 319, 743–748. [DOI] [PubMed] [Google Scholar]
- Butti M. G.; Bongarzone I.; Ferraresi G.; Mondellini P.; Borrello M. G.; Pierotti M. A. A sequence analysis of the genomic regions involved in the rearrangements between TPM3 and NTRK1 genes producing TRK oncogenes in papillary thyroid carcinomas. Genomics 1995, 28, 15–24. [DOI] [PubMed] [Google Scholar]
- Greco A.; Mariani C.; Miranda C.; Pagliardini S.; Pierotti M. A. Characterization of the NTRK1 genomic region involved in chromosomal rearrangements generating TRK oncogenes. Genomics 1993, 18, 397–400. [DOI] [PubMed] [Google Scholar]
- Bounacer A.; Schlumberger M.; Wicker R.; Du-Villard J. A.; Caillou B.; Sarasin A.; Suarez H. G. Search for NTRK1 proto-oncogene rearrangements in human thyroid tumours originated after therapeutic radiation. Br. J. Cancer 2000, 82, 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beimfohr C.; Klugbauer S.; Demidchik E. P.; Lengfelder E.; Rabes H. M. NTRK1 re-arrangement in papillary thyroid carcinomas of children after the Chernobyl reactor accident. Int. J. Cancer 1999, 80, 842–847. [DOI] [PubMed] [Google Scholar]
- Tognon C.; Knezevich S. R.; Huntsman D.; Roskelley C. D.; Melnyk N.; Mathers J. A.; Becker L.; Carneiro F.; MacPherson N.; Horsman D.; Poremba C.; Sorensen P. H. B. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [DOI] [PubMed] [Google Scholar]
- Lannon C. L.; Sorensen P. H. B. ETV6-NTRK3: A chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin. Cancer Biol. 2005, 15, 215–223. [DOI] [PubMed] [Google Scholar]
- Watanabe N.; Kobayashi H.; Hirama T.; Kikuta A.; Koizumi S.; Tsuru T.; Kaneko Y. Cryptic t(12;15)(p13;q26) producing the ETV6-NTRK3 fusion gene and no loss of IGF2 imprinting in congenital mesoblastic nephroma with trisomy 11 fluorescence in situ hybridization and IGF2 allelic expression analysis. Cancer Genet. Cytogenet. 2002, 136, 10–16. [DOI] [PubMed] [Google Scholar]
- Izbicka E.; Izbicki T. Therapeutic strategies for the treatment of neuroblastoma. Curr. Opin. Invest. Drugs 2005, 6, 1200–1214. [PubMed] [Google Scholar]
- Douma S.; Van Laar T.; Zevenhoven J.; Meuwissen R.; Van Garderen E.; Peeper D. S. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TRKB. Nature 2004, 430, 1034–1039. [DOI] [PubMed] [Google Scholar]
- Miknyoczki S. J.; Chang H.; Klein-Szanto A.; Dionne C. A.; Ruggeri B. A. The TRK tyrosine kinase inhibitor CEP-701 (KT-5555) exhibits significant antitumor efficacy in preclinical xenograft models of human pancreatic ductal adenocarcinoma. Clin. Cancer Res. 1999, 5, 2205–2212. [PubMed] [Google Scholar]
- Ketterer K.; Rao S.; Friess H.; Weiss J.; Buechler M. W.; Korc M. Reverse transcription-PCR analysis of laser-captured cells points to potential paracrine and autocrine actions of neurotrophins in pancreatic cancer. Clin. Cancer Res. 2003, 9, 5127–5136. [PubMed] [Google Scholar]
- Sclabas G. M.; Fujioka S.; Schmidt C.; Li Z.; Frederick W. A. I.; Yang W.; Yokoi K.; Evans D. B.; Abbruzzese J. L.; Hess K. R.; Zhang W.; Fidler I. J.; Chiao P. Overexpression of tropomysin-related kinase B in metastatic human pancreatic cancer cells. Clin. Cancer Res. 2005, 11, 440–449. [PubMed] [Google Scholar]
- Wang T.; Yu D.; Lamb M. L. TRK kinase inhibitors as new treatments for cancer and pain. Expert Opin. Ther. Patents 2009, 19, 305–319and references therein. [DOI] [PubMed] [Google Scholar]
- Bouhana K. S.; Impastato R.; Pheneger J.; Jiang Y.; Wallace R. D.; Do M. G.; Zautke N. A.; Andrews S. W.. Analgesic Effects of a Potent and Selective Kinase Inhibitor of Neurotrophin Receptors TRKA, TRKB and TRKC. www.arraybiopharma.com; WO2010033941; WO2010048314; WO11006074.
- Okram B; Nagle A.; Adrian F. J.; Lee C.; Ren P.; Wang X.; Sim T.; Xie Y.; Wang X.; Xia G.; Spraggon G; Warmuth M.; Liu Yi.; Gray N. S. A General Strategy for Creating ‘‘Inactive-Conformation’’ Abl Inhibitors. Chem. Biol. 2006, 13, 779–786. [DOI] [PubMed] [Google Scholar]
- Melnick J. S.; Janes J.; Kim S.; Chang J. Y.; Sipes D. G.; Gunderson D.; Jarnes L.; Matzen J. T.; Garcia M. E.; Hood T. L.; Beigi R.; Xia G.; Harig R. A.; Asatryan H.; Yan S. F.; Zhou Y.; Gu X. J.; Saadat A.; Zhou V.; King F. J.; Shaw C. M.; Su A. I.; Downs R.; Gray N. S.; Schultz P. G.; Warmuth M.; Caldwell J. S. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 3153–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Structure coordinates have been deposited with the PDB: code 3V5Q.
- Dar A. C.; Lopez M. S.; Shokat K. M. Small Molecule Recognition of c-Src via the Imatinib-Binding Conformation. Chem. Biol. 2008, 15, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard J. R.; Klueter S.; Gruetter C.; Getlik M.; Rabiller M.; Rode H. B.; Rauh D. A. New screening assay for allosteric inhibitors of cSrc. Nat. Chem Biol. 2009, 5, 394–396. [DOI] [PubMed] [Google Scholar]
- Pargellis C; Tong L.; Churchill L.; Cirillo P. F.; Gilmore T.; Graham A. G.; Grob P. M.; Hickey E. R.; Moss N.; Pav S.; Regan J. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [DOI] [PubMed] [Google Scholar]
- Wan P. T. C.; Garnett M. J.; Roe S. M.; Lee S.; Niculescu-Duvaz D.; Good V. M.; Jones C. M.; Marshall C. J.; Springer C. J.; Barford D.; Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [DOI] [PubMed] [Google Scholar]
- PD data for compound 22 (GNF-5837) will be described in detail in a manuscript currently in preparation and to be published elsewhere.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.