

Abstract
Guided by computational methods, a set of 1920 compounds were selected from the AstraZeneca corporate collection and screened for Kv1.5 activity. To facilitate rapid generation of structure–activity relationships, special attention was given to selecting subsets of structurally similar molecules by using a maximum common substructure similarity-based procedure. The focused screen hit rate was relatively high (12%). More importantly, a structural series featured by the symmetric 1,2-diphenylethane-1,2-diamine substructure was identified as potent Kv.1.5 blockers. The property profile for the series is shown to meet stringent lead-optimization criteria, providing a springboard for the development of a new and safe treatment for atrial fibrillation.
Keywords: atrial fibrillation, Kv1.5 blockers, focused screen, maximum common substructure, structural series, symmetric ligands
Atrial fibrillation is the most commonly sustained cardiac arrhythmia in clinical practice and is implicated in approximately 15% of all strokes in the United States.1 A promising therapeutic approach is to selectively block the ultrarapid delayed rectifier potassium ion current (IKur) and thereby delay atrial repolarization and convert atrial fibrillation to normal sinus rhythm.2 The voltage-gated potassium Kv1.5 ion channel underlies IKur, and its functional activity is predominantly found in the human atrium.3 By blocking IKur, an atrial-selective increase in action potential duration and effective refractory period (ERP) is understood to be achieved, which in turn can decrease atrial fibrillation inducibility.4 The discovery of structurally diverse Kv1.5 blockers was recently reviewed by Bilodeau and Trotter.5 Some representative compounds that inhibit the Kv1.5 channel are shown in Figure 1.
Figure 1.
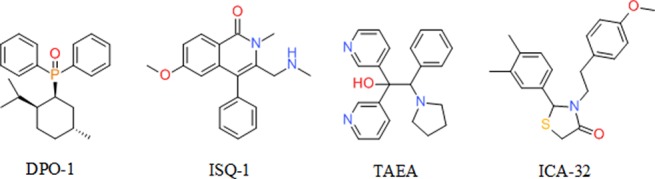
Molecular structures of potent Kv1.5 blockers: DPO-1,6 ISQ-1,7 TAEA,8 and ICA-32.9
To provide an existing Kv1.5 lead optimization (LO) program with series of new, potent structures having favorable physicochemical properties, a lead generation (LG) back-up program was initiated. Many different approaches can be used to find attractive leads to start a drug discovery project.10 High-throughput screening (HTS) of large corporate compound collections is one approach typically used, but it is arguably not always necessary.11 When extensive knowledge of the target and active ligands is available, approaches such as focused screening are often superior alternatives.11 We therefore decided to opt for the more cost- and time-efficient virtual screens (VS), allowing for testing in the existing high-quality low-throughput LO screening assays. In short, the chosen approach was a limited focused screen guided by computational methods.
Peukert et al. have described use of various virtual screening methods as successful approaches to identify Kv1.5 blockers.12,13 Their approaches may be viewed as “compound-centric”, whereas our strategy may be described as “structural series-centric”. During virtual screening, little attention is typically made to arrive at series of compounds, and the number of near-neighbors in a cluster is seldom monitored. As a result, such focused screens may contain unnecessarily large clusters and an unreasonable large fraction of singletons (Figure 2a).
Figure 2.
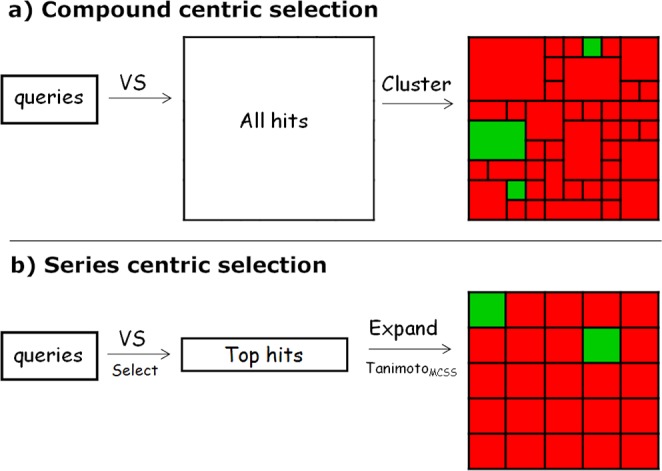
(a) Commonly used compound-centric virtual screening strategy may result in the selection of many singletons and big clusters, as visualized schematically by the different box sizes. The series-centric strategy (b), on the other hand, focuses on expanding the top VS hits into a more even distribution of compounds per series to allow for a speedier LG phase.
In an attempt to alleviate the uncertainty associated with obtaining singletons as hits and to ensure the economical use of limited screening resources (e.g., avoid screening big clusters), special attention was given to selecting series of similar compounds (Figure 2b). Twenty compounds were arbitrarily chosen as the “optimal” cluster size for a structural series, anticipating grounds for informed decision making. A potential advantage of this strategy is that the sets of near-neighbors are especially suitable for deriving structure–activity relationships. Another benefit is that that the number of screen iterations can be kept to a minimum, leading to a shorter LG phase. On the other hand, active compounds with very few near-neighbors will by definition not be discovered using this strategy, and the approach may not be optimal for difficult targets where low hit rates are expected.
Rather than depending on one particular computational method, a wide variety of methods were used in the initial VS: fingerprints,14 shape- and electrostatic complementarities,15,16 lingo similarities,17 and maximum common substructure (MCS)-based techniques.18 The results from each method will depend on the query structures, and the results are typically complementary.19 The query structures were carefully selected from relevant sources (e.g., our internal LO program, literature and competitor patents). The VSs were performed on the AstraZeneca corporate collection, filtered according to typical drug-likeness criteria.20 In addition, compounds registered with low purity (<85%) and compounds containing known toxophores or reactive groups were removed to provide high-quality hit structures to allow for an efficient LG phase.
The 500 highest ranked compounds from each VS search method were visually inspected and assessed in relation to the parent query. General physicochemical descriptors, with an extra emphasis on selecting compounds with low lipophilicity, and medicinal chemistry knowledge were used to select the most attractive starting points—top hits. The top hits thus selected were subjected to MCS-based similarity searches to expand the hits into structural series of near-neighbors. The method used was a MCS-based similarity measure (TanimotoMCSS) calculated using the OpenEye OEChem-toolkit,15 according to eq 1.18
| 1 |
where NA and NB are the number of atoms in molecules A and B, respectively, and NAB is the number of atoms in the MCSS of A and B. A TanimotoMCSS value of 1 is obtained if two molecules are identical. The reason for using a substructure-based similarity measure was to ensure that core features of the hit molecules were preserved and that only molecules with small modifications were retrieved. Such small modifications have a strong correspondence with the type of functional group replacement common in medicinal chemistry projects. This is in contrast to fingerprint-based similarity comparisons where the structural similarity features are not always straightforward to rationalize and thus produce clusters that look unnatural to a medicinal chemist. As a consequence, key determinants of biological activity may be difficult to pinpoint and even more difficult to exploit in the design of improved analogues.
A final number of 82 structural series were selected, with an average cluster size of 23 compounds, leading to 1920 compounds in total. These compounds were tested for Kv1.5 activity in an Rb+ efflux assay. As many as 230 compounds were defined as actives (>40% inhibition at 10 μM), leading to a hit rate of 12%. The actives obtained from the primary screen were retested for purity (LC/MS) to confirm their molecular identity, and IC50 values were then determined. The great majority (90%) of the actives showed IC50 values less than 30 μM, and 17% showed IC50 values less than 10 μM. Roughly 20 series contained four or more active compounds.
Figure 3 depicts the structures of one of the most interesting hit series. It contained 15 compounds, and the MCS is the 1,2-diphenylethanamine fragment highlighted in blue. This particular structural series was identified using the MCS similarity-based search and the competitor compound 16 (1-[2-(cyclopropylmethoxy)-1,2-diphenylethyl]pyridin-2-one)21 as a query, to provide the top hit (15). Compound 15 was subsequently expanded into the structural series shown in Figure 3. The calculated TanimotoMCSS between the top VS hit 15 and the query 16 is relatively high (0.63). There were only a handful of compounds in the AstraZeneca corporate collection with higher values at the time. The TanimotoMCSS for the remaining compounds in the 1,2-diphenylethanamine series as calculated from the query 16 end up in the mean distribution, meaning that they were not “top VS hits”. On the other hand, compounds 1–14 are all structural near-neighbors of the top hit 15, with TanimotoMCSS ≥ 0.63.
Figure 3.
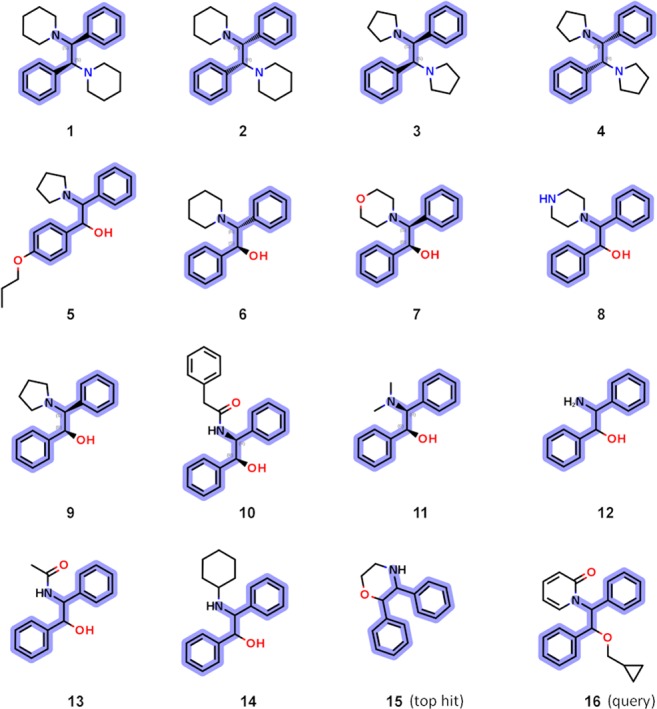
VS using 16 as the query resulted in the top hit compound 15, which in turn was expanded into the structural series shown. The MCS for the series is the 1,2-diphenylethanamine as highlighted in blue. Compounds 1–4 were found to be potent Kv1.5 blockers.
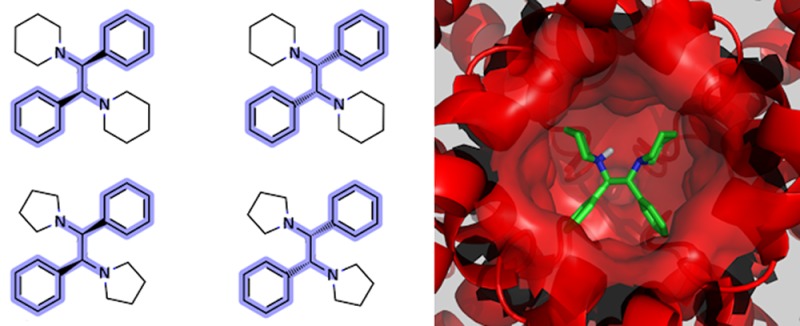
The four 1,2-diphenylethane-1,2-diamine stereoisomers (1–2 and 3–4)22,23 showed high affinity (0.2–0.6 μM) for the Kv1.5 channel and are equipotent with the more lipophilic reference compound DPO-1 (Kv1.5 IC50 = 0.5 μM, and log D = 6.0). The remaining compounds 5–15 were found to be inactive. The Kv1.5 IC50 values were determined by electrophysiological studies using the high-throughput planar patch clamp assay and IonWorks described by Schroeder et al.24
From a structural perspective, compounds 1–4 are atypical since they are symmetric. Some medicinal chemists have prejudices against symmetric compounds.25 One common argument is that high symmetry will lead to stable crystals and thus low solubility. Another argument is that binding sites are rarely symmetrical. Nevertheless, there are many symmetric drugs in clinical use,26 and in this case, symmetric ligands seem to be in harmony with the 3D structure of the target protein. There is currently no experimentally determined crystal structure of the Kv1.5 ion channel, but the complete Kv1.2 channel−β2 subunit complex structure has been determined, revealing a highly symmetric tetramer (PDB entry: 2A79).27 The sequence identity between Kv1.2 and Kv1.5 is very high, and importantly, the amino acids in the pore domains are highly conserved (90%), allowing for the construction of a Kv1.5 homology model.28 Figure 4 displays a Kv1.5 homology model based on the Kv1.2 crystal structure, proposing a conceivable ligand binding mode for compound 4. The high shape complementarity between the protein and the docked ligand is notable.
Figure 4.
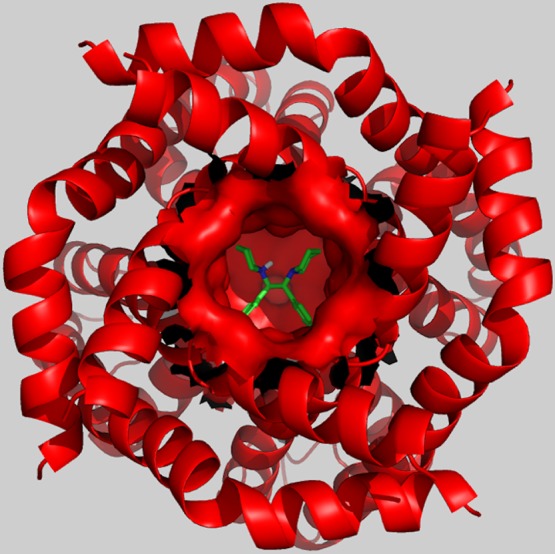
Symmetric compound 1 docked in a homology model of the Kv1.5 ion channel illustrating the high shape complementarity between protein and ligand.
Unlike previously reported potent Kv1.5 blockers, compounds 1–4 include an ionizable functional group. The presence of a basic nitrogen flanked by aromatic moieties prompted us to test the compounds for hERG inhibition, as many ligands that block the hERG channel are known to contain this motif.29 From a regulatory perspective, there is a general requirement for selectivity against binding to the hERG channel, and this is particularly true for new and safe antiarrythythmics. Figure 5 shows the measured hERG inhibition data, and compounds 1–4 were fortunately found to be inactive, exhibiting IC50 values above the highest assay concentration (>33 μM) and thus reducing our concern for cardiac arrhythmic side effects.
Figure 5.

Summary of the properties of the novel series of 1,2-diphenylethane-1,2-diamine Kv1.5 blockers. The boxes are color-coded to monitor the distance from the target profile, where the green color denotes that the particular compound fulfills that particular project criterion for the next milestone transition.
Furthermore, the above-mentioned pharmacophore of the 1,2-diphenylethane-1,2-diamine compounds 1–4 is sometimes associated with inhibition of one common cytochrome P450 enzyme: the 2D6 subtype.30 Consequently, the compounds were tested for CYP2D6 inhibition, and submicromolar IC50 values were discovered (Figure 5). This information was included in the subsequent LO strategy with the goal of eliminating any such effect to increase metabolic stability and to minimize the possible effect of drug–drug interactions.
In addition to the already mentioned experiments, the diamine structural series, together with a small number of other interesting series, were exposed to extensive profiling. That is, several parameters frequently used for decision-making in drug discovery were determined; see Figure 5. For example, the four representative compounds (1–4) were found to be potent in the secondary Kv1.5 voltage clamp assay. Most other parameters, such as aqueous solubility, reactive metabolite formation, and CaCO-2 permeability, were found to be well within our defined thresholds. Moreover, in vivo pharmacokinetic studies were conducted in rat, providing promising results. Figure 5 summarizes the experimentally determined properties of the novel diamine series and shows the corresponding criteria for LO transfer. Such tables may be used to aid decision making at milestone transitions, as they simplify risk identification, as well as allow easy comparison between lead series.
In summary, a limited focused screening effort guided by computational methods, using a number of known Kv1.5 blockers as starting points, resulted in a novel series of symmetric Kv1.5 blockers with favorable properties in a timely fashion. The time between LG start and having the necessary data to demonstrate that the stringent LO criteria were met was a relatively short 9 months. This provides support for the use of focused screening as the method of choice when the aim is to identify new series for existing LO projects. The strategy of selecting series instead of compounds was successful in this case. In addition, feedback that the structural series were considered to be well-defined clusters of near-neighbors was received. On a more general note, selecting series based on substructures is very much aligned to the way medicinal chemists perceive SAR. Other benefits of selecting series based on substructures and using defined quality criteria are that it enables an appreciation of the series feasibility and its optimization potential and assesses any risks associated with it. The results of further medicinal chemistry work on the 1,2-diphenylethane-1,2-diamine structural series will be reported in the near future.
Acknowledgments
Fanyi Jiang is acknowledged for running the Kv1.5 experiments and the many other colleagues at AstraZeneca CVGI Mölndal for their helpful assistance in generating data as well as for helpful discussions.
Glossary
Abbreviations
- HTS
high-throughput screen
- VS
virtual screen
- MCS
maximum common substructure
- LG
lead generation
- LO
lead optimization
- ERP
effective refractory period
- IKur
ultrarapid delayed rectifier potassium ion current
- Kv1.5
voltage-gated potassium (Kv) channel subunit 5
Supporting Information Available
Coordinates for the homology model and descriptions of the experimental methods used to measure Kv1.5 IC50, log D, solubility, hERG inhibition, pKa values, CYP inhibition, and HLM Clint. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Lip G. Y. H; Tse H.-F. Management of atrial fibrillation. Lancet 2007, 370, 604–618. [DOI] [PubMed] [Google Scholar]
- Brendel J.; Peukert S. Blockers of the Kv1.5 channel for the treatment of atrial arrhythmias. Curr. Med. Chem. Cardiovasc. Hematol. Agents 2003, 1, 273–287. [DOI] [PubMed] [Google Scholar]
- Feng J. L.; Wible B.; Li G. R.; Wang Z. G.; Nattel S. Antisense oligodeoxynucleotides directed against Kv1.5 mRNA specifically inhibit ultrarapid delayed rectifier K current in cultured adult human atrial myocytes. Circ. Res. 1997, 80, 572–579. [DOI] [PubMed] [Google Scholar]
- Knobloch K.; Brendel J.; Peukert S.; Rosenstein B.; Busch A. E.; Wirth K. J. Electrophysiological and antiarrhythmic effects of the novel IKur channel blockers, S9947 and S20951, on left vs right pig atrium in vivo in comparison with the IKr blockers dofetilide, azimilide, d,l-sotalol and ibutilide. Naunyn Schmiedeberg's Arch. Pharmacol. 2002, 366, 482–487. [DOI] [PubMed] [Google Scholar]
- Bilodeau M. T.; Trotter B. W. Kv1.5 Blockers for the treatment of atrial fibrillation: approaches to optimization of potency and selectivity and translation to in vivo pharmacology. Curr. Top. Med. Chem. 2009, 9, 436–451. [DOI] [PubMed] [Google Scholar]
- Lagrutta A.; Wang J.; Fermini B.; Salata J. J. Novel, potent inhibitors of human Kv1.5 K+ channels and ultra-rapidly activating delayed rectifier potassium current. J. Pharmacol. Exp. Ther. 2006, 317, 1054–1063. [DOI] [PubMed] [Google Scholar]
- Trotter B. W.; Nanda K. K.; Kett N. R.; Regan C. P.; Lynch J. J.; Stump G. L.; Kiss L.; Wang J.; Spencer R. H.; Kane S. A.; White R. B.; Zhang R. N.; Anderson K. D.; Liverton N. J.; McIntyre C. J.; Beshore D. C.; Hartman G. D.; Dinsmore C. J. Design and synthesis of novel isoquinoline-3-nitriles as orally bioavailable Kv1.5 antagonists for the treatment of atrial fibrillation. J. Med. Chem. 2006, 49, 6954–6957. [DOI] [PubMed] [Google Scholar]
- Regan C. P.; Kiss L.; Stump G. L.; McIntyre C. J.; Beshore D. C.; Liverton N. J.; Dinsmore C. J.; Lynch J. J. Atrial antifibrillatory effects of structurally distinct I-Kur blockers 3-[(Dimethylamino)methyl]-6-methoxy-2-methyl-4-phenylisoquinolin-1(2H)-one and 2-phenyl-1,1-dipyridin-3-yl-2-pyrrolidin-1-yl-ethanol in dogs with underlying heart failure. J. Pharmacol. Exp. Ther. 2008, 324, 322–330. [DOI] [PubMed] [Google Scholar]
- Wu S.; Fluxe A.; Sheffer J.; Janusz J. M.; Blass B. E.; White R.; Jackson C.; Hedges R.; Murawsky M.; Fang B.; Fadayel G. M.; Hare M.; Djandjighian L. Discovery and in vitro/in vivo studies of tetrazole derivatives as Kv1.5 blockers. Bioorg. Med. Chem. Lett. 2006, 16, 6213–6218. [DOI] [PubMed] [Google Scholar]
- Valler M. J.; Green D. Diversity screening versus focussed screening in drug discovery. Drug Discovery Today 2000, 5, 286–293. [DOI] [PubMed] [Google Scholar]
- Schnecke V.; Boström J. Computational chemistry-driven decision making in lead generation. Drug Discovery Today 2006, 11, 43–50. [DOI] [PubMed] [Google Scholar]
- Peukert S.; Brendel J.; Pirard B.; Strübing C.; Kleemann H.-W.; Böhme T.; Hemmerle H. Pharmacophore-based search, synthesis, and biological evaluation of anthranilic amides as novel blockers of the Kv1.5 channel. Bioorg. Med. Chem. Lett. 2004, 14, 2823–2827. [DOI] [PubMed] [Google Scholar]
- Pirard B.; Brendel J.; Peukert S. The discovery of Kv1.5 Blockers as a case study for the application of virtual screening approaches. J. Chem. Inf. Model. 2005, 45, 477–485. [DOI] [PubMed] [Google Scholar]
- Blomberg N.; Cosgrove D. A.; Kenny P W.; Kolmodin K. Design of compound libraries for fragment screening. J. Comput.-Aided Mol. Des. 2009, 23, 513–525. [DOI] [PubMed] [Google Scholar]
- OEChem Tool-kit, OpenEye Scientific Software, Santa Fe, NM; http://www.eyesopen.com/oechem-tk; accessed May 25, 2012.
- ROCS, OpenEye Scientific Software, Santa Fe, NM; http://www.eyesopen.com./rocs; accessed May 25, 2012.
- Grant J. A.; Haigh J. A.; Pickup B. T.; Nicholls A.; Sayle R. A. Lingos, finite state machines, and fast similarity searching. J. Chem. Inf. Model. 2006, 46, 1912–1918. [DOI] [PubMed] [Google Scholar]
- Boström J.; Hogner A.; Schmitt S. Do structurally similar molecules bind in a similar fashion. J. Med. Chem. 2006, 49, 6716–6725. [DOI] [PubMed] [Google Scholar]
- Sheridan R. P.; Kearsley S. K. Why do we need so many chemical similarity search methods?. Drug Discovery Today 2002, 7, 903–911. [DOI] [PubMed] [Google Scholar]
- Lipinski C. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- Brendel J.; Englert H. C.; Wirth K.; Wagner M.; Ruxer J.-M.; Pilorge F.. Substituted heterocycles, their use as medicament, and pharmaceutical preparations comprising them. WO-2006/136304, 2006.
- Hatano B.; Tachikawa T.; Mori T.; Nagahashi K.; Kijima T. Reductive coupling of aromatic N,N-acetals using zinc and chlorotrimethylsilane. Tetrahedron Lett. 2011, 52, 3467–3469. [Google Scholar]
- Betschart C.; Seebach D. Preparation of 1,2-diarylethylenediamines by aminative reductive coupling of aromatic aldehydes with low-valent titanium reagents. Helv. Chim. Acta 1987, 70, 2215–2231. [Google Scholar]
- Schroeder K.; Neagle B.; Trezise D. J.; Worley J. IonWorksTM HT: A new high-throughput electrophysiology measurement platform. J. Biomol. Screening 2003, 8, 50–64. [DOI] [PubMed] [Google Scholar]
- “In the Pipeline” blog post accessed May 25, 2012; http://pipeline.corante.com/archives/2008/10/20/fearful_symmetry.php.
- A selection of symmetrical drugs in clinical use are nitroglycerin, suramin, dipyridamole, ethambutol, Felbamate, Pentamidine, Mitoxantrone and pentaerythritol tetranitrate.
- Long S. B.; Campbell E. B.; MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [DOI] [PubMed] [Google Scholar]
- Ander M.; Luzhkov V. B.; Åqvist J. Ligand binding to the voltage-gated Kv1.5 potassium channel in the open state—Docking and computer simulations of a homology model. Biophys. J. 2008, 94, 820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli A.; Poluzzi E.; De Ponti F.; Recanatini M. Toward a pharmacophore for drugs inducing the long QT syndrome: Insights from a CoMFA study of HERG K+ channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Ekins S. Pharmacophore modeling of cytochromes P450. Adv. Drug Delivery Rev. 2002, 54, 367–383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.