

Abstract

The discovery of new Bcl-2 protein–protein interaction antagonists is described. We replaced the northern fragment of ABT737 (π–π stacking interactions) with structurally simplified hydrophobic cage structures with much reduced conformational flexibility and rotational freedom. The binding mode of the compounds was elucidated by X-ray crystallography, and the compounds showed excellent oral bioavailability and clearance in rat PK studies.
Keywords: Bcl-2 inhibitors, protein−protein interaction, π−π stacking interactions, π−π stacking isosteres, bioisoteres, cancer therapeutics
Although the principles and energetic components of biomolecular binding interactions have been well studied, the design and implementation of these principles remain a challenge. Nowhere is this challenge more pronounced than in the search for small molecule inhibitors of protein–protein interactions (PPI’s).1 A notable recent success was achieved with the discovery of ABT737,2−4 a dual Bcl-2, Bcl-xL inhibitor with potent in vivo activity against a number of tumors.5,6 Bcl-2 and Bcl-xL are representative members of the B-cell lymphoma-2 (Bcl-2) family of antiapoptotic proteins, which also include Bcl-w, A1, and Mcl-1.7 The overexpression of these proteins in cancer cells is a defining event in chemo- and radiotherapy resistance, and it also marks the progression of the disease. Mechanistically, the antiapoptotic Bcl-2 family members prevent the programmed cell death by binding/sequestering the pro-apoptotic Bcl-2 family members (e.g., bid, bim, noxa). This sequestration prevents the oligomerization of Bax/Bak, which is essential for the permeation of the mitochondrial membrane, subsequent release of cytochrome C, and initiation of downstream apoptosis events.8−12
Inspection of the binding interactions between ABT737 and Bcl-2 reveals a highly optimized ligand (with respect to the protein–ligand interactions) spanning the p2 to p4 binding pockets.13 Interestingly, these two pockets were independently identified by alanine-scanning mutagenesis as being critical “hot spots” in the interaction of Bcl-2 with Bak.14 Notable among the interactions is a unique turn in the northern fragment of ABT737 where the thiophenyl is folded underneath the nitroaromatic ring (intramolecular π–π stacking interaction) with the latter ring forming an additional intermolecular π–π stacking with Tyr 161 of Bcl-2. This folding of the ligand in the p4 pocket has three negative consequences: First, the electronic demand of the π–π interactions restricts the medicinal chemistry approaches to improving the druglike properties of ABT737.15 Second, the highly engineered nature of the p4 ligand portion of ABT737 introduces a large amount of rotational freedom and lipophilicity to the molecule. High levels of rotatable bonds and lipophilicity have a generally negative impact on solubility and permeability. Lastly, the folding itself would entail significant conformational changes. It is difficult to accurately quantify the energetics of these conformational changes, but it is generally accepted that they will largely negatively impact the binding potency.16 Thus, reducing the conformational flexibility of ABT737 represents a viable approach to improving not only its druglike properties but perhaps also its binding potency.17,18 Efforts along this line have been reported in the literature.19 Herein, using ABT737 as a starting point, we describe an orthogonal design principle for developing new Bcl-2 inhibitors with significantly reduced conformational flexibility. Efforts to improve the solubility of this novel series are also discussed.
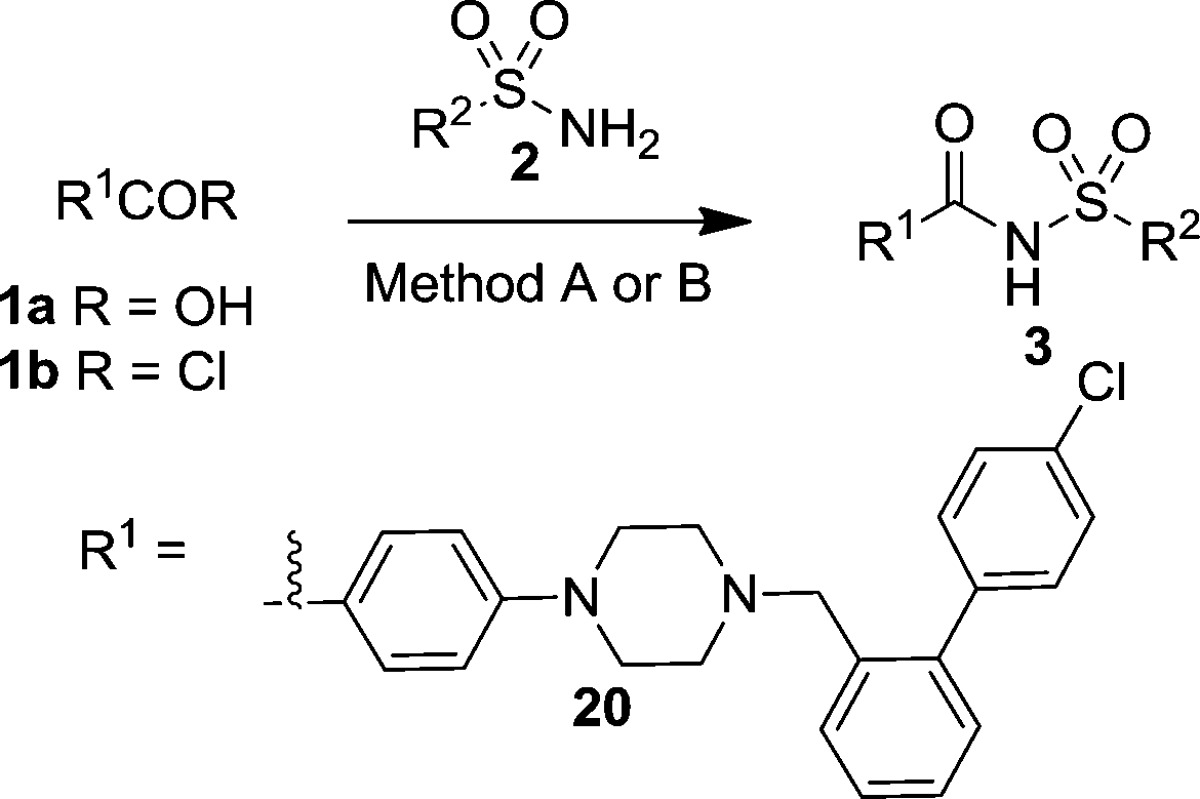
At the outset, we elected to maintain the chlorophenyl and linker regions (piperazine to sulfone) constant, as they make several important interactions with the protein.20 Thus, our efforts started with compound 3a, wherein the entire northern ABT737 fragment is replaced with a simple methyl group. Remarkably, measurable binding interactions remained with an EC50 of 8.62 μM (Table 1, entry 1).21 Sequential homologation of this methyl group quickly exposed the promises and challenges of our approach. For instance, while 3b was more potent (5.31 μM, Table 1), the potency was reduced when the methyl group was replaced with t-Bu (3c, Table 1, entry 3), indicative of steric clashes with the protein. In agreement, 3d was also inactive. Despite these setbacks, we continued to prepare bulkier analogues such as 3e and 3f; this time we also inserted a methylene linker between the sulfonamide functionality and the p4 probe. We were pleased to observe that 3e showed a near 8-fold improvement in potency compared to the case of 3a. Moreover, enantiomeric cis-myrtanol derived analogues 3g and 3h, lacking any polar atoms, displayed significantly improved inhibition of the target (0.56 and 0.42 μM, respectively). Again, similar to the cases of the camphor derivatives (vide supra), no stereodiscrimination was observed in the binding.
Table 1. Redesigning the Northern Fragment of ABT737.

Average of at least two measurements, the compounds are inactive against Mcl-1 (IC50 > 50 μM).
Stereochemistry of the starting camphor sulfonamides.
Enantiomeric (1S,2S,5S)- and (1R,2R,5R)-myrtanol were used respectively to prepare these analogues.
This is the bottom limit of the assay.
With a good understanding of the binding requirements in the p4 pocket, we revisited the adamantane analogue 3d. Its appeal stems from its high degree of symmetry and relative synthetic tractability. The synthesis of 3i, featuring a two-carbon linker, is outlined in Scheme 1.22,23 First, the commercially available primary alcohol 5 was treated with triphenylphosphine and iodine to afford iodide 6, which was immediately displaced with potassium thioacetate to yield intermediate 7 in quantitative yield. The oxidation of this newly formed thioacetate 7 with sulfuryl chloride and subsequent treatment with ammonium hydroxide allowed rapid access to 8 in 71% yield. Further oxidation of 8 into sulfonamide 9 using potassium permanganate paved the way for the final coupling step as shown in Table 1 to afford 3i. 3j was also prepared in an analogous fashion. Gratifyingly, both compounds were active against the target, with 3j being a sub-micromolar inhibitor of Bcl-2 (Table 1, entry 8). These results further highlight the necessity for a linker with one carbon being the optimal length. The nature of this spacer was also critical. For instance, the conversion of the carbon linker into amine (sulfonyl urea, synthesis described in Scheme 2) negatively impacted the potency (3k, Table 1).
Scheme 1. Conversion of Alcohols into Sulfonamides.

Conditions: (a) l2, PPh3, imidazole, DCM, 15 min, rt; (b) AcSK, THF, 60 °C; (c) Ac2O, SO2Cl2, DCE, −10 °C, 30 min, rt, 30 min, then NH4OH; (d) aq KMnO4, acetone.
Scheme 2. Synthesis of the Sulfonyl Urea.

Conditions: (a) t-BuOH, DCM, rt, 10 min, then adamantylamine, 0 °C to rt, 45 min; (b) HCl, dioxane.
Having optimized the linker, we now turned our attention to the p4 binding region of the molecule. Again, sharp SAR trends were manifest. The contraction of the adamantane ring by one carbon resulted in 4-fold loss in potency (3l vs 3j, Table 1). Leaving this region of the molecule intact, we inserted an additional carbon into the distal position in the adamantane framework (3m, Table 1). Remarkably, this expansion of the ring by a single carbon atom resulted in a net 6-fold gain in potency. These results demonstrate how potency may be modulated in the series. We also tested key compounds against Bcl-xL, a closely related Bcl-2 family member. In all cases (compounds 3h, 3j, and 3m, Table 1), the compounds were marginally less active, with 3j providing the largest window of selectivity (4.3-fold).
At this juncture, we sought to solve the crystal structure of some of our potent Bcl-2 binders to validate our design hypothesis and inform the SAR optimization strategy. This campaign led to the X-ray complex of 3j and Bcl-2 (Figure 1a). It shows that the adamantane is occupying the hydrophobic p4 pocket per the initial design assumptions. The adamantane ring fits within the binding envelope defined by the northern region of ABT737, although a close inspection shows movement of Tyr 161, a key residue for the binding of ABT737 (not shown). This movement creates a deeper hydrophobic pocket to accommodate the binding of the ligand. Other notable differences between the two crystal structures are highlighted in Figure 1b. The acylsulfonamide moiety (3j) is rotated away from the protein. This repositioning of the sulfonamide was also observed in the X-ray cocomplex of 3a, a less sterically demanding analogue, and Bcl-2. From this we infer that the position of the sulfonamide found in the ABT737 structure is being repositioned by the π–π stacking motif that forces the sulfonamide from its lowest energy positioning. Contrast this with the high symmetric adamantyl group which imposes much less bias on the sulfonamide position. More importantly, this rotation results in the loss of a hydrogen bonding interaction between the sulfone oxygen (3j) and the backbone NH of Gly104 in Bcl-2 (this interaction is seen in the X-ray cocomplex of ABT737 and Bcl-2). The above results clearly validated our design premise, and demonstrated for the first time that the northern region of ABT737 can be replaced with simple rigid structural motifs with much reduced rotational freedom.
Figure 1.

Crystal structures. (a) X-ray complex of 3j and Bcl-2 at 2.0 Å resolution (left). (b) Overlay of the X-ray crystal structures of ABT-737 and compound 3j (right).
Guided by this cocrystal structure, we next attempted to access the region occupied by the dimethyl amine moiety in the crystal structure of the ABT737/Bcl-2 complex as well as the subpocket utilized by the nitro group (ABT737). An additional goal of these efforts was to decrease the clogP of our compounds, to improve the solubility of our analogues. The synthesis of these analogues is detailed in Scheme 3. It started with 3-bromoadamantane-1-carboxylic acid (12). First, reduction of the carboxylic acid with BH3·THF afforded alcohol 13 in excellent yield (87%). The latter compound was converted into 14 via four straightforward and high yielding synthetic operations as described above in Scheme 1. This versatile intermediate lent itself to many interesting chemistries. For instance, the angular Br can be displaced with aqueous phosphate under microwave conditions to afford alcohol 15 in 71% yield. Further, 16 was obtained in good yield when the latter compound was reacted with DAST in DCM at room temperature. Additionally, treatment of 14 with alcohols, exemplified by 2-morpholinoethanol, under forcing conditions afforded alkoxyadamantane analogues in moderate yield. Finally, bromide 14 can be carried through the Ritter reaction with a variety of nitrile coupling partners to afford the corresponding N-adamantylamides in good yield (18, Scheme 3). These intermediates, as well other commercially available 3-functionalized adamantane methyl sulfonamides, were then successfully coupled to the bottom ABT737 carboxylic acid (20) to afford the desired final targets 19a–d and 19g–j (Table 2). The 3-position of the adamantane could also be probed via a late stage diversification strategy. For instance, the fluoride in 19c was replaced with a methyl group by simple treatment with trimethyl aluminum (19e, Scheme 3).
Scheme 3. Derivatization of Adamantane Core.

Conditions: (a) BH3·THF, THF, 0 °C, then reflux, 4 h; (b) see Scheme 1; (c) aq NaH2PO4, microwave, 180 °C, 10 min; (d) DAST, DCM, 24 h; (e) 2-morpholinoethanol, microwave, 200 °C, 90 min; (f) RCN, ZnCl2 (1 M, in Et2O), DCE, 90 °C, 12 h; (g) 20, EDCI, DMF/DCE; (h) Me3Al, DCM, rt, 30 min.
Table 2. Modulation of the Druglike Properties and Potency in the Adamantane Series.


Average of at least two measurements (duplicate EC50 values were generated in each assay).
Number of rotatable bonds. For comparison ABT737 has 17 rotatable bonds and a clogP of 10.
The potency of these second generation analogues was measured in the SPR assay, and the results are summarized in Table 2. Interestingly, hydroxylation of the adamantane at the 3-position was not only tolerated but also afforded a more potent compound (19a, Table 2). Furthermore, sterically larger polar substituents such as the side chain ethyl morpholinyl (19b, Table 2) were also tolerated at this position.
However, 19a and 19b were equipotent, hinting that the latter compound binds orienting the 3- position away from the pocket (solvent exposed). In agreement with this data, hydrophobic substituents at this position were not tolerated. For instance, a simple methylation (19e) resulted in a 2-fold loss in potency. Furthermore, the introduction of a phenyl ring (19f) was even less favorable. In contrast, halogens (fluoro (19c), bromo (19d)) were tolerated. Finally, while smaller hydrophobic amide substituents were tolerated, and even improved the potency of our series in some instances (19g–i), bigger hydrophobic amides were less desirable (19i). Overall, we concluded that this position may be ideally used for the introduction of solubilizing groups, and this goal can be combined with minor gains in potency as shown for the hydroxyl and smaller amide analogues.
The design concept of introducing a simple rigid structure to replace the northern portion of ABT737 was intended to improve the overall pharmaceutical properties of our Bcl-2 inhibitors by reducing MW, clogP, and rotatable bonds (the clogP and number of rotatable bonds values are shown in Table 2 for our compounds24). The pharmacokinetic properties obtained for compounds 3j and 19a after a single solution dose in Sprague–Dawley rat are summarized in Table 3. The systemic clearance values were low relative to the hepatic blood flow (6 and 16 mL/min·kg, respectively). The volumes of the distributions were moderate (3j, 0.7 L/kg; 19a, 1.2 L/kg), with the drug being primarily confined to the plasma. Both compounds showed excellent oral bioavailability (66 and 100%25), indicative of improved permeability and solubility relative to the case of ABT737. Overall, the pharmacokinetic properties of both compounds were favorable.
Table 3. In Vivo Pharmacokinetics in SpraguesDawley Rata.
| parameters | 3j | 19a |
|---|---|---|
| Cl (mL/min·kg) | 6 | 16 |
| Vdss (L/kg) | 0.7 | 1.2 |
| t1/2 (h) | 1.3 | 1.3 |
| %F | 66 | >100b |
| clogP | 9.5 | 6.4 |
Sprague–Dawley rat was dosed 1 mg/kg iv and 10 mg/kg po. The compound was administered as a solution in 10% NMP, 30% PEG300, 60 (20%) vitamin ETPGS for 3j and in 10% NMP, 25% PEG300, 5% cremophor EF,WFI for 19a.
See related comments in ref (25).
In summary, we have demonstrated that simple and rigid cage structures can replace the highly engineered northern fragment in ABT737, albeit with a minimal loss in potency. Herein, the binding is primarily driven by hydrophobic interactions, but we have identified areas of the molecules that can be modified to improve the solubility of the series. More importantly, a favorable PK profile is obtained in the process as compared to the case of ABT737. Combining good potency and oral exposure has been challenging in targeting Bcl-2, and the results reported herein are very promising in this regard. Further efforts to improve the potency of our compounds while preserving the favorable PK profile are in progress and will be reported in due course.
Supporting Information Available
Synthetic procedures and characterization data (1H NMR, HRMS, and HPLC analysis) for compounds 3a–3m, 19a–e, and g–j, and biochemical assay conditions. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Keskin O.; Gursoy A.; Ma B.; Nussinov R. Principles of Protein–Protein Interactions: What are the Preferred Ways For Proteins To Interact?. Chem. Rev. 2008, 108, 1225–1244. [DOI] [PubMed] [Google Scholar]
- Petros A. M.; Dinges J.; Augeri D. J.; Baumeister S. A.; Betebenner D. A.; Bures M. G.; Elmore S. W.; Hajduk P. J.; Joseph M. K.; Landis S. K.; Nettesheim D. G.; Rosenberg S. H.; Shen W.; Thomas S.; Wang X.; Zanze I.; Zhang H.; Fesik S. W. Discovery of a Potent Inhibitor of the Antiapoptotic Protein Bcl-xL from NMR and Parallel Synthesis. J. Med. Chem. 2006, 49, 656–663. [DOI] [PubMed] [Google Scholar]
- Wendt W. D. Discovery of ABT-263, a Bcl-family protein inhibitor: observations on targeting a large protein–protein interaction. Expert Opin. Drug Discovery 2008, 3, 1123–1143. [DOI] [PubMed] [Google Scholar]
- Park C.-M.; Bruncko M.; Adickes J.; Bauch J.; Ding H.; Kunzer A.; Kennan M. C.; Nimmer P.; Shoemaker A. R.; Song X.; Tahir S. K.; Tse C.; Wang X.; Wendt M. D.; Yang X.; Zhang H.; Fesik S. W.; Rosenberg S. H.; Elmore S. Discovery of an Orally Bioavailable Small Molecule Inhibitor of Prosurvival B-Cell Lymphoma 2 Proteins. J. Med. Chem. 2008, 51, 6902–6915. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T.; Elmore S. W.; Shoemaker A. R.; Armstrong R. C.; Augeri S. W.; Belli B. A.; Bruncko M.; Deckwerth T. L.; Dinges J.; Hajduk P. J.; Joseph M. K.; Kitada S.; Korsmeyer S. J.; Kunzer A. R.; Letai A.; Li C.; Mitten M. J.; Nettesheim D. J.; Ng S.; Nimmer P. M.; O’Connor J. M.; Oleksijew A.; Petros A. M.; Reed J. C.; Shen W.; Tahir S. K.; Thompson C. B.; Tomaselli K. J.; Wang B.; Wendt M. D.; Zhang H.; Fesik S. W.; Rosenberg S. H. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [DOI] [PubMed] [Google Scholar]
- Tse C.; Shoemaker A. R.; Adickes J.; Anderson M. G.; Chen J.; Jin S.; Johnson E. F.; Marsh K. C.; Mitten M. J.; Nimmer P.; Roberts L.; Tahir S. K.; Xiao Y.; Yang X.; Zhang H.; Fesik S.; Rosenberg S. H.; Elmore S. W. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- Cory S.; Adams J. M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Danial N. N.; Korsmeyer S. J. Cell death: critical control points. Cell 2004, 116, 205–219. [DOI] [PubMed] [Google Scholar]
- For Bcl-2 overexpression in cancers see:; Berghella A. M.; Pellegrini P.; Contasta I.; Del Beato T.; Adorno D. Bcl-2 and Drugs Used in the Treatment of Cancer: New Strategies of Biotherapy which should not be Underestimated. Cancer Biother. Radiopharm. 1998, 13, 225–236. [DOI] [PubMed] [Google Scholar]
- For correlation between Bcl-2 and resistance to treatment with radiation and chemotherapy see:; Reed J. C. Mechanisms of apoptosis avoidance in cancer. Curr. Opin. Oncol. 1999, 11, 68–75. [DOI] [PubMed] [Google Scholar]
- Kusenda J. Bcl-2 family proteins and leukemia. Neoplasma (Bratisl.) 1998, 45, 117–122. [PubMed] [Google Scholar]
- Lee E. F.; Czabotar P. E.; Smith B. J.; Deshayes K.; Zobel K.; Colman P. M.; Fairlie W. D. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007, 14, 1711–1713. [DOI] [PubMed] [Google Scholar]
- Sattler. M.; Liang H.; Nettesheim D.; Meadows R. P.; Harlan J. E.; Eberstadt M.; Yoon H. S.; Shuker S. B.; Chang B. S.; Minn A. J.; Thompson C. B.; Fesik S. W. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 1997, 275, 983–986. [DOI] [PubMed] [Google Scholar]
- Lipinsky C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Koshland D. The active site and enzyme action. Adv. Enzymol. Relat. Subj. Biochem. 1960, 22, 45–97. [DOI] [PubMed] [Google Scholar]
- Bartlett P. A.; Yusuff N.; Pyun H.-J.; Rico A. C.; Meyer J. H.; Smith W. W.; Burger M. T. Design of Macrocyclic Peptidase Inhibitors: The Related Roles of Structure based Approaches and Library Chemistry. In Medicinal Chemistry into the Millenium. Spec. Publ.—R. Soc. Chem. 2001, 264, 3–15. [Google Scholar]
- Hruby V. J. Design in topographical space of peptide and peptidomimetic ligands that affect behavior. A chemist’s glimpse at the mind–body problem. Acc. Chem. Res. 2001, 34, 389–397. [DOI] [PubMed] [Google Scholar]
- Park C.-M.; Oie T.; Petros A. M.; Zhang H.; Nimmer P. M.; Henry R. F.; Elmore S. W. Design, Synthesis, and Computational Studies of Inhibitors of Bcl-XL. J. Am. Chem. Soc. 2006, 128, 16206–16212. [DOI] [PubMed] [Google Scholar]
- Goreshnik I.; Brock A. M.; Maly D. J. Biochemical and pharmacological profiling of the pro-survival protein Bcl-xL. Bioorg. Med. Chem. Lett. 2011, 21, 4951–4955. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for a description of this assay format.
- Toshikazu K.; Yuichi I.; Matsubara H.; Hobara D.; Kakiuchi T.; Okazaki T.; Komatsu K. Rigid Molecular Tripod with an Adamantane Framework and Thiol Legs. Synthesis and Observation of an Ordered Monolayer on Au(111). J. Org. Chem. 2006, 71, 1362–1369. [DOI] [PubMed] [Google Scholar]
- Thea S.; Cevasco G. A facile synthesis of sulfinyl chlorides from thiolacetates. Tetrahedron Lett. 1987, 28, 5193–5194. [Google Scholar]
- In comparison, ABT737 has a clogP of 10.0 and 17 rotatable bonds.
- The oral bioavailability values were respectively 97, 108, and 150% in individual rats. Saturation of clearance at higher po relative to IV dose may explain the >100% F in the third rat.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.