

Abstract

On the basis of scaffold hopping, a novel series of 2-alkyl-1-arylsulfonylprolinamides was discovered as 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD-1) inhibitors. A representative compound 4ek, obtained through SAR and structure optimization studies, demonstrates excellent in vitro potency against 11β-HSD-1 and dose-dependent in vivo inhibition of 11β-HSD-1 in a prednisone/prednisolone transformation biomarker study in mice.
Keywords: metabolic syndrome, enzyme inhibitor, 11β-hydroxysteroid dehydrogenase type 1, sulfonamide, 2-alkylproline, prolinamide
Glucocorticoid receptor (GR) signaling plays a significant role in metabolic regulation, and defects in this signaling pathway have been implicated in the development of several phenotypes of metabolic syndrome.1 GR signaling depends not only on the circulating cortisol levels but also on the intracellular production of cortisol through reduction of cortisone, the inactive glucocorticoid. The enzymes catalyzing the conversion between cortisone and cortisol are 11β-hydroxysteroid dehydrogenases (11β-HSDs). Among them, the type 1 isoform (11β-HSD-1), highly expressed in liver and adipose tissue, predominantly reduces cortisone to cortisol, and the type 2 isoform (11β-HSD-2), primarily expressed in kidney, oxidizes cortisol to cortisone. A potential role for 11β-HSD-1 inhibitors in metabolic syndrome, type 2 diabetes, and obesity has been established using transgenic mice.2−2c On the basis of these findings, in recent years, 11β-HSD-1 is recognized as a promising target in metabolic disease.1−2c
In the past decade, industrial and academic researchers have reported several classes of 11β-HSD-1 inhibitors with varied scaffolds, including sulfonamides (e.g., BVT-14225, 1a), amides (e.g., PF-877423, 2) (Figure 1), triazoles (e.g., Merck 544), and thiazolones (e.g., AMG-221).3−3d In a phase II clinical trial, 11β-HSD-1 inhibitor INCB-13739 (structure undisclosed) significantly improved insulin sensitivity in type 2 diabetes patients who failed on metformin treatment and lowered triglyceride and cholesterol levels of patients with hyperlipidaemia and hypertriglyceridemia.4 Currently, other examples such as PF-915275, MK-0916, and AZD-4071 are being evaluated in phase I/II trials for potential oral treatment of metabolic diseases.5−5c
Figure 1.

11β-HSD-1 inhibitors. Combining the arylsulfonyl moiety of 1a or 1b with the d-prolinamide moiety of PF-877423 (2) to discover 2-alkyl-1-arylsulfonylprolinamide 4a.
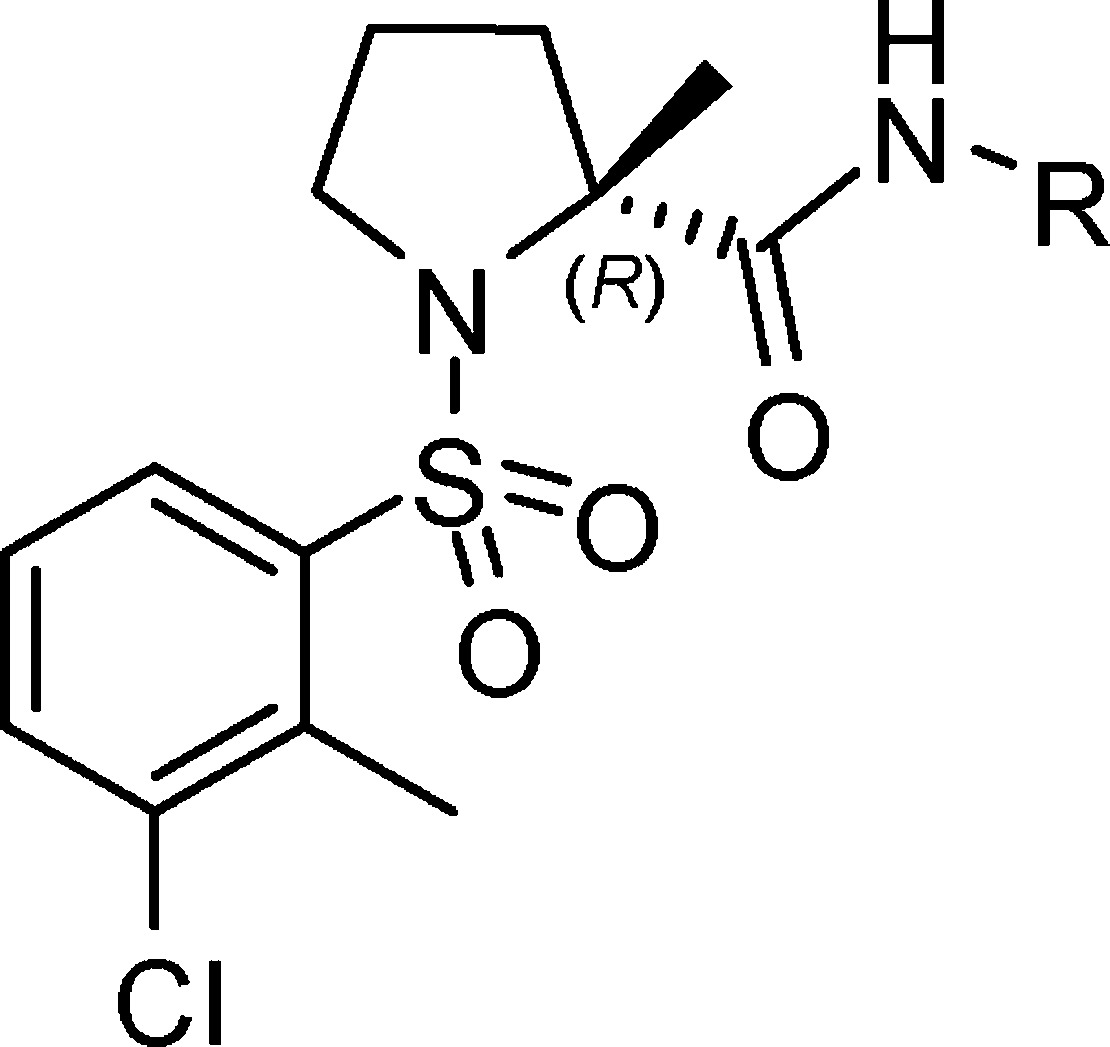
We recently disclosed a new series of sulfonamides (e.g., 1b, Figure 1) with high inhibitory activity against 11β-HSD-1, and compound 1b showed a short duration of action in a pharmacodynamics (PD) model.6 As part of our continuing efforts to find novel and proprietary 11β-HSD-1 inhibitors, we combined the arylsulfonyl moiety of 1a or 1b with d-prolinamide moiety of 2 to form a new sulfonamide-d-prolinamide represented by 3 (Figure 1), which displayed only micromolar activity against human 11β-HSD-1 (h-11β-HSD-1) but poor liver microsome stability (HLM Clint 430 μL/min/mg). In vitro metabolite identification studies indicated that the pyrrolidine ring of compound 3 was oxidized by HLM. We thought the substituent (e.g., alkyl) on the ring might be helpful to slow down the oxidization, so we first selected the commercially available (R)-α-methyl proline as a starting material and obtained compound 4a. Interestingly, 4a exhibited not only better HLM stability but also dramatically increased potency (IC50 = 77 nM). Utilizing 4a as a starting point, we discovered and elaborated a series of 2-alkyl-1-arylsulfonylprolinamides as potent and selective 11β-HSD-1 inhibitors. Herein, we report the synthesis, structure–activity relationship (SAR), and optimization of this series.
2-Alkyl-1-arylsulfonylprolinamide derivatives have been prepared according to Schemes 1–3. The inhibitory activity of this series of compounds against h-11β-HSD-1 and mouse 11β-HSD-1 (m-11β-HSD-1) was determined by scintillation proximity assay (SPA) method,6−7b and the results are listed in Tables 1–5. The selectivity over human 11β-HSD-2 (h-11β-HSD-2) and 3T3L1 cellular activity of some representative compounds was also tested.
Scheme 1. Synthesis of Compounds 4a–g and 8a,b,e.

Reagents and conditions: (a) CH3OH, SOCl2, reflux, 1 h. (b) 3-Chloro-2-methylbenzene-1-sulfonyl chloride, triethylamine, CH2Cl2, rt, 2 h. (c) NaOH, THF, CH3OH, H2O, rt, overnight. (d) RNH2, BOP-Cl, DIPEA, CH2Cl2, rt, 6 h.
Scheme 3. Synthesis of Compounds 17a–c.

Reagents and conditions: (a) Triethylamine, CH2Cl2, rt, 2 h. (b) TMS-CF3, TBAF, THF, overnight. (c) NaOH, CH3OH, THF, H2O, rt, overnight. (d) 4-Aminoadamantane, BOP-Cl, DIPEA, CH2Cl2, 6 h.
Table 1. In Vitro Potency of (R)-2-Methyl Prolinamides with Varied Amino Groups.
Average of at least two replicates.
Percent inhibition at 100 nM, the mean of at least two experiments.
Table 5. In Vitro Potency of Compounds 18a,b.

| IC50a (nM) |
|||
|---|---|---|---|
| compd | R2 | h-11β-HSD-1 | m-11β-HSD-1 |
| 4ek | –CH3 | 7.7 | 11 |
| 18a | –CH2CH3 | 16 | 164 |
| 18b | –CH2CH=CH2 | 33%b | 87 |
Average of at least two replicates.
Percent inhibition at 100 nM, the mean of at least two experiments.
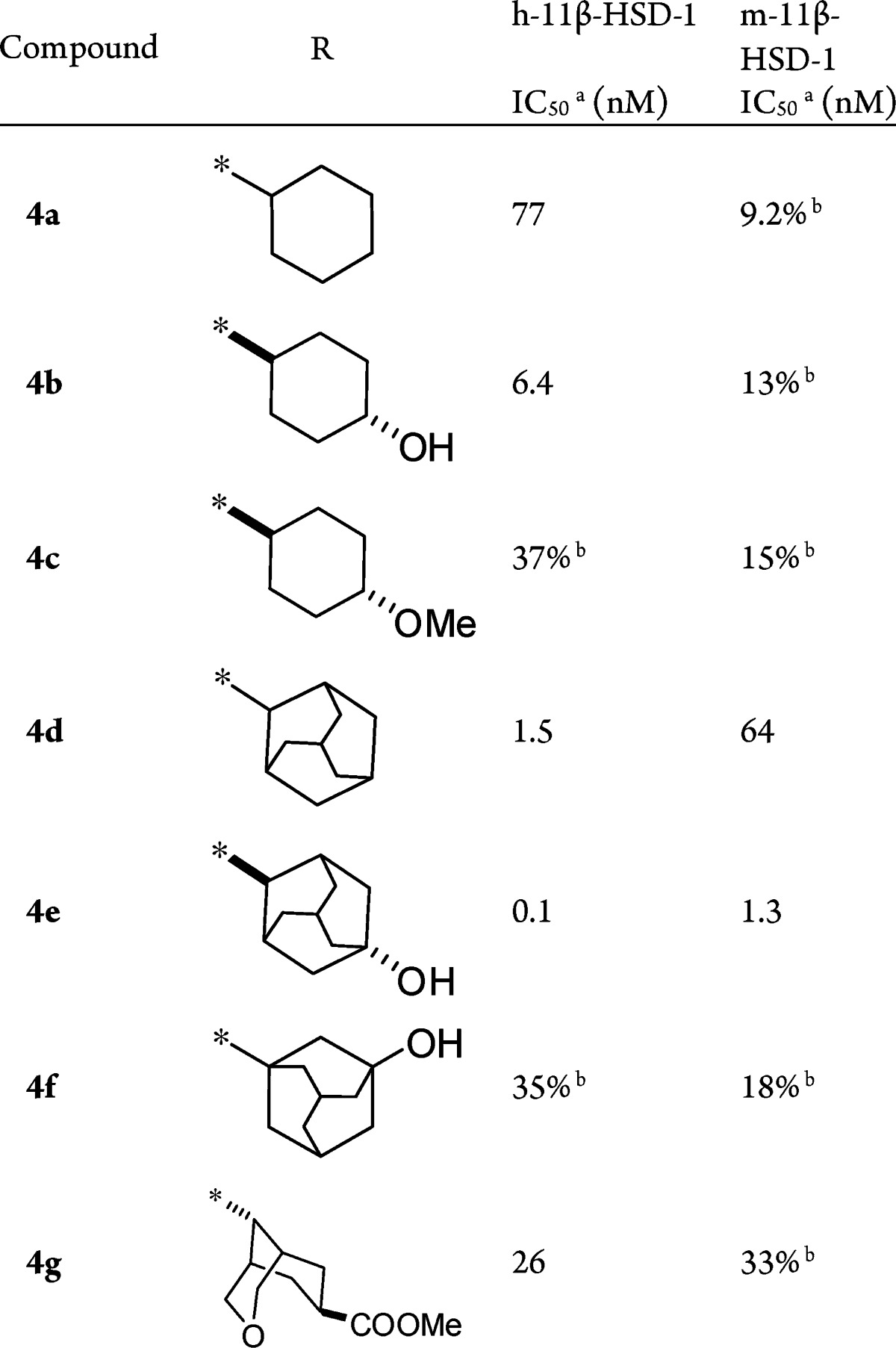
As seen in Scheme 1, esterification of (R)-2-methylpyrrolidine-2-carboxylic acid (5) gave (R)-methyl 2-methylpyrrolidine-2-carboxylate (6). Typical sulfonylation of 6 and saponification of the resulting ester produced carboxylic acid 7, followed by coupling with varied primary amines using bis(2-oxo-3-oxazolidinyl)phosphonic chloride (BOP-Cl) to yield target molecules 4a–g. As summarized in Table 1, when a hydroxyl group is introduced to the 4-position of the cyclohexyl moiety, the potency toward h-11β-HSD-1 increased 12-fold (4b vs 4a); however, the methoxyl derivative 4c exhibited reduced activity (37% at a concentration of 100 nM), and all three compounds appeared to have very weak inhibition toward m-11β-HSD-1 (no more than 15% at a concentration of 100 nM). The adamant-2-yl-substituted 4d and 4e showed single-digit nanomolar and subnanomolar inhibition against human enzyme (IC50 = 1.5 and 0.1 nM); moreover, they were also potent inhibitors of rodent enzyme with IC50 values of 64 and 1.3 nM, respectively. However, other bridged hydrocarbon substituents such as adamant-1-yl (4f) and 3-oxabicyclo[3.3.1]nonane (4g) led to poor activity versus rodent enzyme.
For comparison, using (S)-acid 9 instead of (R)-acid 5, we synthesized (S)-form derivatives 8a, 8b, and 8e (Scheme 1). As shown in Table 2, although the (S)-derivatives had lower activity than the corresponding (R)-derivatives 4a, 4b, and 4e, the adamant-2-amino analogue 8e exhibited high activity toward both human and rodent 11β-HSD-1.
Table 2. In Vitro Potency of (S)-Enantiomers.


Average of at least two replicates; ND, not determined.
Because h-11β-HSD-1 enzyme shares only about 79% homology with the mouse enzyme, it might be understandable that many 11β-HSD-1 inhibitors displayed species-dependent activity.3a Some clinical candidates, such as INCB-13739 and PF-915275, lack potency against rodent enzyme, so in vivo pharmacokinetics (PK)/PD studies of these compounds require primate models.8 In our series, cross-species potencies of adamant-2-yl derivatives 4d and 4e allowed us to employ rodent model.
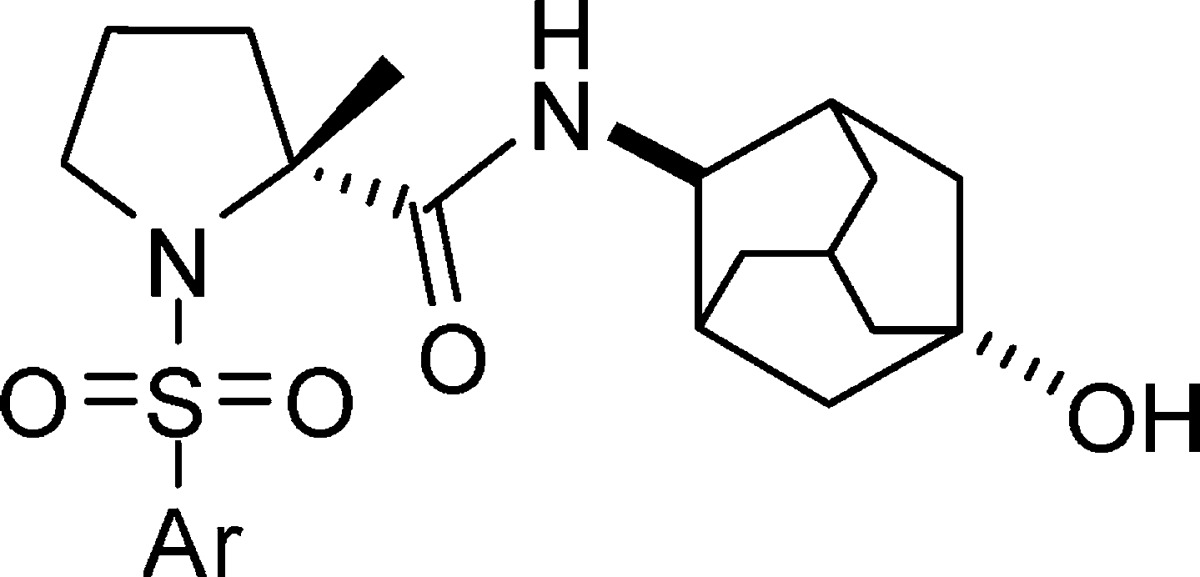
It is reported that the adamantane group is prone to oxidation in living organism, which could cause a rapid elimination of the compound, so substituted adamantanes have been used to prepare metabolically stable druglike molecules.9−9c For example, introducing a hydroxyl group on the adamantane ring reduced Clog P and blocks metabolic soft spots.3b In our test, hydroxylated adamant-2-yl derivative 4e possessed better stability in HLM than unsubstituted adamant-2-yl derivative 4d (HLM Clint of 4e vs 4d: 106 vs 499 μL/min/mg protein). Consequently, we next focused our attention on the derivatization of 4e.
Through the route depicted in Scheme 2, we synthesized more 4-amino-1-hydroxyl-adamantyl derivatives. (R)-1-(tert-Butoxycarbonyl)-2-methyl pyrrolidine-2-carboxylic acid 12, obtained from (R)-acid 5, coupled with trans-4-aminoadamantan-1-ol hydrochloride to provide intermediate 13. The target compounds (4eb–el) were then obtained by two successive steps from 13: deprotection and sulfonylation. As listed in Table 3, most of the aryl or heteroaryl groups maintained high binding affinities to human enzyme with single-digit nanomolar IC50 values, but some of them lose rodent enzyme activity.
Scheme 2. Synthesis of Compounds 4eb–el.

Reagents and conditions: (a) (Boc)2O, Na2CO3, dioxane, H2O, rt, 0.5–2 h. (b) trans-4-Aminoadamantan-1-ol hydrochloride, BOP-Cl, DIPEA, CH2Cl2, rt, overnight. (c) CF3COOH, CH2Cl2, rt, 1–6 h. (d) ArSO2Cl, DIPEA, CH2Cl2, rt, 2–16 h.
Table 3. In Vitro Potency and Liver Microsomal Stability of (R)-2-Methyl Prolinamides with Varied Arylsulfonyl Groups.

Average of at least two replicates.
Percent inhibition at 100 nM, the mean of at least two experiments; ND, not determined.
To extend the SAR of this series, we next explored the effect of the R1 group on adamantane. Noticing 4-(1,1,1-trifluoro-2-hydroxypropan-2-yl)benzenesulfonamide reported in literature as a pharmacophore of 11β-HSD-1 inhibitors, we designed new derivatives 17a–c (Scheme 3 and Table 4).10 As described in Scheme 3, sulfonylation of 6 afforded ketone 15. Intermediate 16 was generated by treatment of 15 with (trifluoromethyl)trimethylsilane (TMS-CF3)/tetra-n-butylammonium fluoride (TBAF) in THF and a following hydrolysis step. The target compounds were then produced via condensation of 16 with the 1-substituted 4-aminoadamantanes. As listed in Table 4, the potency against human enzyme of 17b (R1 = CN) or 17c (R1 = COOMe) decreases 7- or 20-fold as 17a (R1 = OH). However, all three compounds display only weak activity on rodent enzyme at a concentration of 100 nM.
Table 4. In Vitro Potency of Compounds 17a–c.
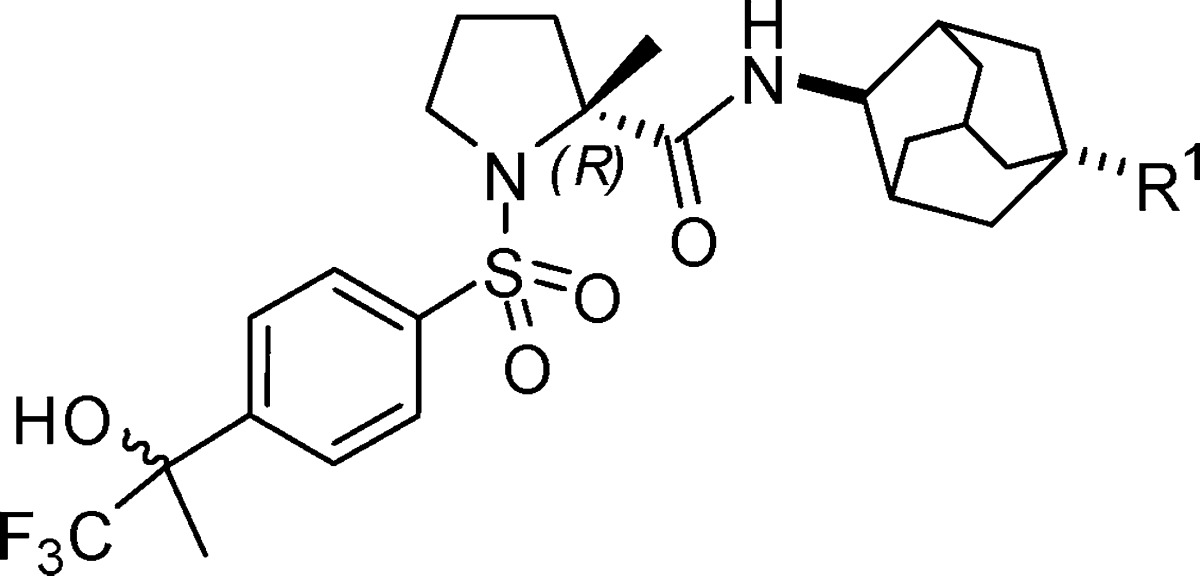
| compd | R1 | h-11β-HSD-1 IC50a (nM) | m-11β-HSD-1 inh. at 100 nMb (%) |
|---|---|---|---|
| 17a | OH | 6.0 | 26 |
| 17b | CN | 43 | 9.5 |
| 17c | COOMe | 124 | 14 |
Average of at least two replicates.
The mean of at least two experiments.
To investigate the effect of R2 group on the pyrrolidine, 4ek analogues 18a and 18b were designed (Table 5). The synthesis of 18a and 18b is analogous to 4ek, replacing starting material 5 in Scheme 1 with (R)-2-ethyl and (S)-2-ally pyrrolidine-2-carboxylic acid.11 As seen in Table 5, the methyl in the 2-position of pyrrolidine is superior to ethyl and allyl groups in terms of potency, but the 2-allyl analogue 18b has higher affinity to rodent enzyme than human enzyme.
The potential for compounds 4e and 4ek to complete lead candidate criteria was further evaluated (Table 6). Both compounds were potent inhibitors of human and mouse 11β-HSD-1 and highly selective against human 11β-HSD-2. In the 3T3L1 cell-based assay, compound 4ek was 11 times more potent than 4e. In the mouse PK test, however, both 4e and 4ek showed high clearance, very short plasma half-life, and low oral bioavailability.
Table 6. Profile of the Two Lead Compounds.
| 4e | 4ek | |
|---|---|---|
| in vitro assay | ||
| h-11β-HSD-1 IC50a | 0.1 nM | 7.7 nM |
| m-11β-HSD-1 IC50a | 1.3 nM | 11 nM |
| h-HSD-2 IC50a | 2.2 μM | >10 μM |
| selectivity ratio: h-HSD-2/h-11β-HSD-1 IC50 | 22000 | >1300 |
| m-11β-HSD-1 3T3L1 cellular IC50a | 121 nM | 11 nM |
| mice PKb | ||
| CL [mL/(min kg)] | 90 | 60 |
| Vdss (L/kg) | 0.51 | 0.63 |
| iv t1/2 (min) | 14 | 14 |
| po F (%) | 9.1% | 3.0% |
| ip F (%) | 63% | 33% |
| ip AUC0-t (ng h/mL) | 597.0 | 939.0 |
Average of at least two replicates.
Dosed iv (1 mg/kg), po (5 mg/kg), and ip (5 mg/kg) in male BALB/c mice; mean values over three mice.
To look at the correlation between in vitro activity and in vivo inhibition, 4ek, with higher cellular level activity than that of 4e, was selected as a tool compound and progressed to an in vivo 11β-HSD-1 inhibition assay in mice. Because of the poor metabolic stability and low oral bioavailability of 4ek, we at first selected intraperitoneal (ip) injection as administration route (Table 6, enhanced bioavailability after ip injection is 33%, 11 times of the po bioavailability). In this mechanistic biomarker study, normal mice were dosed with 4ek (or vehicle) and then exogenous substrate prednisone, which transformed to prednisolone catalyzed by 11β-HSD1.12 As illustrated in Figure 2, ip treatment of BALB/c mice with 3, 10, and 30 mg/kg 4ek dose- dependently reduced the generation of predinisolone with inhibition of 34, 71, and 89%, respectively, and the corresponding plasma concentrations of 4ek in mice were 28, 80, and 94 ng/mL.
Figure 2.

In vivo inhibitory activity of 11β-HSD-1 in BALB/c mice by 4ek. Student's t test was used to compare the differences between the dosing group and the vehicle. *P < 0.05 vs vehicle.
In conclusion, we reported a new series of 2-alkyl-1-arylsulfonylprolinamides as human and rodent 11β-HSD-1 inhibitors. Primary optimization and SAR studies led to the discovering of compound 4ek, which demonstrated potent and selective activity in vitro and in vivo inhibitory activity in a prednisone conversion experiment. Efforts to improve the PK profile of this series are under way and will be reported in the future.
Acknowledgments
We are indebted to Prof. Ying Leng and Yu Shen, SIMM, for the molecular and cellular level 11β-HSDs assay.
Glossary
Abbreviations
- HSD
hydroxysteroid dehydrogenase
- SAR
structure–activity relationship
- HLM
human liver microsome
- PK
pharmacokinetics
- PD
pharmacodynamics
- BOP-Cl
bis(2-oxo-3-oxazolidinyl)phosphonic chloride
- TMS-CF3
(trifluoromethyl)trimethylsilane
- TBAF
tetra-n-butylammonium fluoride
- DIPEA
N,N-diisopropylethylamine
Supporting Information Available
Experimental procedures and characterization of new chemical entities. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contributed equally.
This work was supported by New Drug Creation Project of the National Science and Technology Major Foundation of China (project 2010ZX09401-404).
The authors declare no competing financial interest.
Supplementary Material
References
- Wang M. Inhibitors of 11β-hydroxysteroid dehydrogenase type 1 in antidiabetic therapy. Handb. Exp. Pharmacol. 2011, 203, 127–146. [DOI] [PubMed] [Google Scholar]
- Saiah E. The role of 11beta-hydroxysteroid dehydrogenase in metabolic disease and therapeutic potential of 11beta-hsd1 inhibitors. Curr. Med. Chem. 2008, 15, 642–649. [DOI] [PubMed] [Google Scholar]
- Masuzaki H.; Yamamoto H.; Kenyon C. J.; Elmquist J. K.; Morton N. M.; Paterson J. M.; Shinyama H.; Sharp M. G.; Fleming S.; Mullins J. J.; Seckl J. R.; Flier J. S. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J. Clin. Invest. 2003, 112, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H.; Bok J. H.; Lee J. H.; Kim I. H.; Kwon S. W.; Lee G. B.; Kang S. K.; Park J. S.; Jung W. H.; Kim H. Y.; Rhee S. D.; Ahn S. H.; Bae M. A.; Ha D. C.; Kim K. Y.; Ahn J. H. Synthesis and biological evaluation of cyclic sulfamide derivatives as 11β-hydroxysteroid dehydrogenase 1 inhibitors. ACS Med. Chem. Lett. 2012, 3, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barf T.; Vallgårda J.; Emond R.; Haeggstroem C.; Kurz G.; Nygren A.; Larwood V.; Mosialou E.; Axelsson K.; Olsson R.; Engblom L.; Edling N.; Roenquist-Nii Y.; Oehman B.; Alberts P.; Abrahmsen L. Arylsulfonamidothiazoles as a new class of potential antidiabetic drugs. Discovery of potent and selective inhibitors of the 11beta-hydroxysteroid dehydrogenase type 1. J. Med. Chem. 2002, 45, 3813–3815. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Hoffman J.; Le P; Nair S. K.; Cripps S.; Matthews J.; Smith C.; Yang M.; Kupchinsky S.; Dress K.; Edwards M.; Cole B.; Walters E.; Loh C.; Ermolieff J.; Fanjul A.; Bhat G. B.; Herrera J.; Pauly T.; Hosea N.; Paderes G.; Rejto P. The development and SAR of pyrrolidine carboxamide 11beta-HSD1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 2897–2902. [DOI] [PubMed] [Google Scholar]
- Hermanowski-Vosatka A.; Balkovec J. M.; Cheng K.; Chen H. Y.; Hernandez M.; Koo G. C.; Le Grand C. B.; Li Z.; Metzger J. M.; Mundt S. S.; Noonan H.; Nunes C. N.; Olson S. H.; Pikounis B.; Ren N.; Robertson N.; Schaeffer J. M.; Shah K.; Springer M. S.; Strack A. M.; Strowski M.; Wu K.; Wu T.; Xiao J.; Zhang B. B.; Wright S. D.; Thieringer R. 11beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J. Exp. Med. 2005, 202, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Véniant M. M.; Hale C.; Hungate R. W.; Gahm K.; Emery M. G.; Jona J.; Joseph S.; Adams J.; Hague A.; Moniz G.; Zhang J.; Bartberger M. D.; Li V.; Syed R.; Jordan S.; Komorowski R.; Chen M. M.; Cupples R.; Kim K. W. St.; Jean D. J.; Johansson L.; Henriksson M. A.; Williams M.; Vallgårda J.; Fotsch C.; Wang M. Discovery of a potent, orally active 11.beta-hydroxysteroid dehydrogenase type 1 inhibitor for clinical study: Identification of (S)-2-((1S,2S,4R)-bicyclo[2.2.1]heptan-2-ylamino)-5-isopropyl-5-methylthiazol-4(5H)-one (AMG 221). J. Med. Chem. 2010, 53, 4481–4487. [DOI] [PubMed] [Google Scholar]
- Rosenstock J.; Banarer S.; Fonseca V. A.; Inzucchi S. E.; Sun W.; Yao W.; Hollis G.; Flores R.; Levy R.; Williams W. V.; Seckl J. R.; Huber R. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 2010, 33, 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney R.; Stewart P. M.; Toh M.; Ndongo M. N.; Calle R. A.; Hirshberg B. Modulation of 11beta-hydroxysteroid dehydrogenase (11betaHSD) activity biomarkers and pharmacokinetics of PF-00915275, a selective 11betaHSD1 inhibitor. J. Clin. Endocrinol Metab. 2008, 93, 550–556. [DOI] [PubMed] [Google Scholar]
- Feig P. U.; Shah S.; Hermanowski-Vosatka A.; Plotkin D.; Springer M. S.; Donahue S.; Thach C.; Klein E. J.; Lai E.; Kaufman K. D. Effects of an 11β-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes. Metab. 2011, 13, 498–504. [DOI] [PubMed] [Google Scholar]
- http://www.astrazenecaclinicaltrials.com/other-drug-products/in_development_drugs/AZD4017/.
- Xia G.; Liu L.; Xue M.; Liu H.; Yu J.; Li P.; Chen Q.; Xiong B.; Liu X.; Shen J. Discovery of novel sulfonamides as potent and selective inhibitors against human and mouse 11β-hydroxysteroid dehydrogenase type 1. Mol. Cell. Endocrinol. 2012, 358, 46–52. [DOI] [PubMed] [Google Scholar]
- Mundt S.; Solly K.; Thieringer R.; Hermanowski-Vosatka A. Development and application of a scintillation proximity assay (SPA) for identification of selective inhibitors of 11beta-hydroxysteroid dehydrogenase type 1. Assay Drug Dev. Technol. 2005, 3, 367–375. [DOI] [PubMed] [Google Scholar]
- Xia G.; Xue M.; Liu L.; Yu J.; Liu H.; Li P.; Wang J.; Li Y.; Xiong B.; Shen J. Potent and novel 11β-HSD1 inhibitors identified from shape and docking based virtual screening. Bioorg. Med. Chem. Lett. 2011, 21, 5739–5744. [DOI] [PubMed] [Google Scholar]
- Bhat B. G.; Hosea N.; Fanjul A.; Herrera J.; Chapman J.; Thalacker F.; Stewart P. M.; Rejto P. A. Demonstration of proof of mechanism and pharmacokinetics and pharmacodynamic relationship with 4′-cyano-biphenyl-4-sulfonic acid (6-amino-pyridin-2-yl)-amide (PF-915275), an inhibitor of 11β–hydroxysteroid dehydrogenase type 1, in cynomolgus monkeys. J. Pharmacol. Exp. Ther. 2008, 3241299–305. [DOI] [PubMed] [Google Scholar]
- Vielhauer E. B.; Brinkman J. A.; Naderi G. B.; Burkey B. F.; Dunning B. E.; Prasad K. A.; Mangold B. L.; Russell M. E.; Hughes T. E. 1-[[(3-hydroxy-1-adamantyl)amino]acetyl]-2-cyano-(S)-pyrrolidine: A potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J. Med. Chem. 2003, 46, 2774–2789. [DOI] [PubMed] [Google Scholar]
- Patel J. R.; Shuai Q.; Dinges J.; Winn M.; Pliushchev M.; Fung S.; Monzon K.; Chiou W.; Wang J.; Pan L.; Wagaw S.; Engstrom K.; Kerdesky F. A.; Longenecker K.; Judge R.; Qin W.; Imade H. M.; Stolarik D.; Beno D. W. A.; Brune M.; Chovan L. E.; Sham H. L.; Jacobson P.; Link J. T. Discovery of adamantane ethers as inhibitors of 11β-HSD1: Synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2007, 17, 750–755. [DOI] [PubMed] [Google Scholar]
- Roche D.; Carniato D.; Leriche C.; Lepifre F.; Christmann-Franck S.; Graedler U.; Charon C.; Bozec S.; Doare L.; Schmidlin F.; Lecomte M.; Valeur E. Discovery and structure–activity relationships of pentanedioic acid diamides as potent inhibitors of 11β-hydroxysteroid dehydrogenase type I. Bioorg. Med. Chem. Lett. 2009, 19, 2674–2678. [DOI] [PubMed] [Google Scholar]
- Julian L. D.; Wang Z.; Bostick T.; Caille S.; Choi R; DeGraffenreid M.; Di Y.; He X.; Hungate R. W.; Jaen J. C.; Liu J.; Monshouwer M.; McMinn D.; Rew Y.; Sudom A.; Sun D.; Tu H.; Ursu S.; Walker N.; Yan X.; Ye Q.; Powers J. P. Discovery of novel, potent benzamide inhibitors of 11beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) exhibiting oral activity in an enzyme inhibition ex vivo model. J. Med. Chem. 2008, 51, 3953–3960. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K.; Yamamoto M.; Watanabe Y.; Ohfune Y. Tetrahedron Lett. 2007, 48, 4821–4824. [Google Scholar]
- Male BALB/c mice (18–22 g, SIPPR-BK Lab Animal Co., Ltd.) were administrated by intraperitoneal (ip) injection with vehicle (N-methyl pyrrolidone:32% β-cyclodextrin:normal saline = 1:6:3/V:V:V) or with 3, 10, and 30 mg/kg of 4ek. One hour after the vehicle or drug treatment, mice were dosed by iv injection with 3 mg/kg of prednisone. Venous blood samples were collected from the retinal vein 2 min later. Plasma prednisone, prolnisolone, and 4ek levels were measured using the LC/MS/MS method (Waters Acquity UPLC system coupled to Applied Biosystems/Sciex API 4000 Q-Trap tandem mass spectrometry).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.