

Abstract

We report a series of irreversible transglutaminase 2 inhibitors starting from a known lysine dipeptide bearing an acrylamide warhead. We established new SARs resulting in compounds demonstrating improved potency and better physical and calculated properties. Transglutaminase selectivity profiling and in vitro ADME properties of selected compounds are also reported.
Keywords: plasma stability, polar surface area, acrylamides, celiac disease, in vitro ADME
Tissue transglutaminase 2 (TG2) is a multifunctional enzyme primarily known for its calcium-dependent protein cross-linking activity.1 Less well-studied functions include simple amidase, GTPase, ATPase, and protein disulfide isomerase activities.2−4 TG2 has been characterized in at least three forms, including open,5 closed,6 and an open-inactive form.7 Genetic deletion of TG2 in mice suggests a role for TG2 activity in mitochondrial energy function,8 and its overactivity has been most closely associated with celiac disease and Huntington's disease (HD). In addition, there is growing support for roles in inflammation and cancer.9−12
HD is an autosomal dominant, progressive, neurodegenerative disease that is characterized clinically by motor, cognitive, and behavioral deficits.13 TG2 expression and transglutaminase activity have been shown to be increased in the brains of HD patients,14 and in vitro and in vivo models have implicated TG2 in HD pathophysiology,15−18 although more recent contradictory animal data have appeared.19
The subject of irreversible inhibitors of TG2 has been recently reviewed.20 Our studies have focused on irreversible inhibitors bearing an acrylamide warhead,21,22 during which we became interested in dipeptides A and B(23) (Figure 1) as leads due to their attractive potency and specificity for TG2. In a prior report,21 we established that an excellent correlation exists for several transglutaminase isoforms between the IC50 values using a 30 min compound incubation and the irreversible inhibition constants, kinact/Ki. With this correlation in hand, we relied on the IC50 values to guide our medicinal chemistry effort. We began by benchmarking dipeptide A, resulting in the selectivity profile illustrated in Figure 1. We also resynthesized and tested several compounds from the Marrano paper,23 the results of which are found in the Supporting Information. To summarize, the results confirmed that dipeptide A was one of the most potent TG2 inhibitors from this report. As shown in Figure 1, ADME profiling studies on this compound indicated good solubility, low permeability potentially accompanied by efflux, and rapid metabolism in mouse liver microsomes (mLM). Our goal was to identify a tool molecule from this series for in vivo proof of concept studies in HD, where brain is the target organ. Therefore, we focused on increasing potency, improving the absorption profile, and increasing microsomal stability. Lowering the polar surface area (PSA), the number of hydrogen bond donors, and the number of rotatable bonds were the main tactics employed. The synthesis of new compounds for this report was accomplished using well-known literature methods, the details of which may be found in the Supporting Information.
Figure 1.

Lysine-based irreversible inhibitors of TG2 from Marrano et al.23
Because of its lower molecular weight, lower PSA, and equipotency, we selected compound B from the Marrano paper as our starting point. Our prior reports described a structural biology/computational chemistry-driven approach to structure–activity relationship (SAR) construction;21,22 however, in the present series, we were unable to successfully use these models to explain the known (or new) SAR. We instead focused on an empirical approach based on improvement of calculated properties. As the carboxylic acid was viewed as an impediment to permeability, we began by replacement with a hydroxmethyl (1) or deletion of this group entirely as in 2 (Table 1). While these modifications resulted in approximately 2- and 4-fold losses in potency relative to B, the PSAs of these compounds (88 and 67, respectively) were significantly improved and in a range more compatible with blood–brain barrier (BBB) permeability. In addition, the relatively modest loss in potency suggested that the carboxylate was not making a productive interaction with TG2.
Table 1. TG2 Activity of Carbamate Derivativesa.

| compd no. | R | R′ | PSA (Å2)b | TG2 IC50 ± SD (μM) |
|---|---|---|---|---|
| B | C6H5 | CO2H | 105 | 2.7 ± 0.32 |
| 1 | C6H5 | CH2OH | 88 | 5.4 ± 0.54 |
| 2 | C6H5 | H | 67 | 7.6 ± 0.78 |
| 3a | H | CO2H | 105 | >80 |
| 3b | 2–Cl-C6H4 | CO2H | 105 | 1.9 ± 0.19 |
| 3c | 2-CF3-C6H4 | CO2H | 105 | 3.5 ± 0.62 |
| 3d | 3-CF3-C6H4 | CO2H | 105 | 10 ± 0.028 |
| 3e | 4-CF3-C6H4 | CO2H | 105 | 33 |
| 3f | 4-NO2-C6H4 | CO2H | 148 | 62 |
| 3g | 4–F-C6H4 | CO2H | 105 | 7.9 ± 0.075 |
| 3h | 4-CH3-C6H4 | CO2H | 105 | 14 ± 0.064 |
| 3i | 4-n-Bu-C6H4 | CO2H | 105 | 5.9 ± 0.68 |
| 3j | 4-t-Bu-C6H4 | CO2H | 105 | 21 |
| 3k | 2,6-F,F-C6H3 | CO2H | 105 | 10 ± 3.7 |
| 3l | 1-naphthyl | CO2H | 105 | 14 ± 0.39 |
| 3m | 2-naphthyl | CO2H | 105 | 9.1 ± 1.3 |
Values accompanied by standard deviations were averaged from at least two independent experiments; they were otherwise obtained in a single determination.
PSA calculations were obtained using the Dotmatics mol2image property calculation engine (http://www.dotmatics.com/).
We next established the SAR with respect to the carbamate moiety (Table 1). With B again as a comparator, the methyl carbamate (3a) resulted in a large loss in potency. Turning again to the benzyl carbamate motif, the 2-chloro (3b) was slightly more potent than B, the 2-trifluoromethyl (3c) was equipotent, and the 3- or 4-trifluoromethyl (3d and 3e, respectively) resulted in 3- and 10-fold losses in potency, respectively. Continuing with 4-substituted benzyl carbamates, a 4-nitro substituent, as in 3f, resulted in a 20-fold loss in potency, while the 4-fluoro (3g) was 2–3-fold less potent. A series of 4-alkyl substituents were next examined (3h–j); however, these were also of lower potency relative to B. Similar results were found with the 2,6-difluoro (3k) and with the 1- and 2-naphthyl derivatives 3l and 3m.
After preliminary investigation of the SAR with respect to the carbamate moiety, we chose to keep the benzyl carbamate constant and turned our attention to replacing the carboxylate with a series of tertiary amides (Table 2). Relatively low molecular weight aliphatic amides as exemplified by 4a–h nearly all showed significantly improved potency relative to B, with several compounds showing submicromolar potency. It was noteworthy that this improvement did not require an increase in PSA, bulking up the molecular weight, or the addition of hydrogen bond donors relative to 1. Observing that a more lipophilic pyrrolidine (4i) afforded 2-fold improved potency relative to 4e, we next prepared 4-phenylpiperidine 4j, which had a TG2 IC50 of 0.054 μM, and proved to be one of the more potent compounds from this series. Phenylpiperidine 4j also had a favorable selectivity profile, with IC50 values of 9.3, > 80, and 21 μM against TG1, TG3, and FXIIIa, respectively.
Table 2. TG2 Activity of Tertiary Amide Derivativesa.
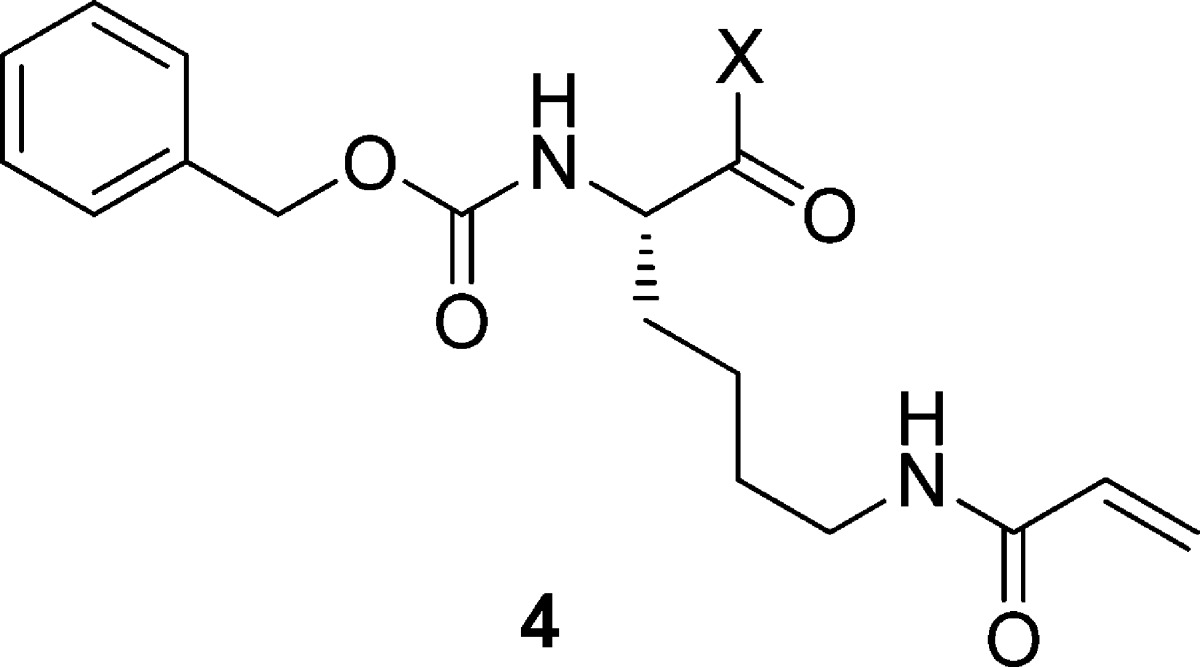

Values accompanied by standard deviations were averaged from at least two independent experiments; they were otherwise obtained in a single determination.
PSA calculations were obtained using the Dotmatics mol2image property calculation engine (http://www.dotmatics.com/).
We followed this with a series of piperazines, which had superior synthetic accessibility facilitating SAR development, but at the cost of increased PSA. Judicious choice of functionality could, in principle, keep the PSA of the piperazines in a more acceptable range. The potency of methylpiperazine 4k (IC50 = 0.61 μM) was in line with the earlier simple tertiary aliphatic amides, indicating that addition of the basic piperazine nitrogen atom was well tolerated. The additional substitution on the terminal phenyl moiety of piperazines 4l and 4m did not affect potency relative to 4j, while a slight loss of potency was observed with 2-naphthyl 4n. Next investigated was a series of 2-piperazinylpyridines (4o–s). As a baseline, pyridine 4o recorded an IC50 of 0.26 μM. The addition of a methyl group at either the 6- (4p) or the 3-position (4q) resulted in improved potency, with 4p having an IC50 of 0.055 μM. Substitution of 4o with a trifluoromethyl group at either the 3- (4r) or the 5-position (4s) resulted in approximately 2-fold improvement in potency relative to 4o. Interestingly, the 3-methyl (4q) and electron-withdrawing 3-trifluoromethyl (4r) and 5-trifluoromethyl (4s) analogues all had similar potency. Favorable transglutaminase selectivity was also retained; 4l had IC50 values of 0.062, 12.9, >80, 69, and 67 μM against TG2, TG1, TG3, TG6, and FXIIIa, respectively. Further profiling also demonstrated that compounds from this series were 2–7-fold less potent against mouse TG2 than they were against the human form (shown in the Supporting Information). This species difference may be an indication of a somewhat different binding mode as compared with our earlier chemotypes,21,22 which did not show this effect.
At this point, we had significantly improved potency while keeping transglutaminase selectivity and the hydrogen bond donor count in favorable ranges. However, the large number of rotatable bonds inherent in the lysine-based scaffold and PSA exceeding 100 for some potent compounds (particularly the 2-piperazinylpyridines) were concerns for oral bioavailability.24 A key tactic employed to reduce the number of rotatable bonds was lysine scaffold replacement. Early attempts resulted in 4-aminophenylalanine-based 5 and piperidinylalanine-based 6 (Figure 2); however, both were inactive against TG2 when tested at 80 μM, and comparisons with the potencies of B, 4p, and 8a argued against synthesis of additional analogues. Improved potency was observed for proline-based 7 (IC50 = 1.8 μM), which was prepared as a diastereomeric mixture. However, given this potency, that it shared many structural features with 4p and would be expected to carry similar liabilities in terms of permeability/efflux and microsomal stability, we elected not to continue the SAR development required to optimize this scaffold.
Figure 2.

Lysine scaffold replacements 5–7.
Another round of SAR development led to the series of 2-piperazinyl-6-methylpyridines shown in Table 3. The bulky Boc derivative 8a was ca. 15-fold less potent than benzyl carbamate 4p. A further loss in activity resulted from incorporation of a 2-naphthylamide (8b). Interestingly, improved potency was seen with acetamide 8c, and an additional improvement was observed from phenylacetamide 8d, which was the most potent compound of the series. Favorable transglutaminase selectivity was also retained; 8d had IC50 values of 0.014, 15, >80, >80, and 35 μM against TG2, TG1, TG3, TG6, and FXIIIa, respectively. Benzamide 8e was equipotent with 8c, demonstrating the importance of the methylene linker in positioning and orienting the phenyl substituent.
Table 3. TG2 Potency of 2-Piperazinyl-6-methylpyridine Derivativesa.
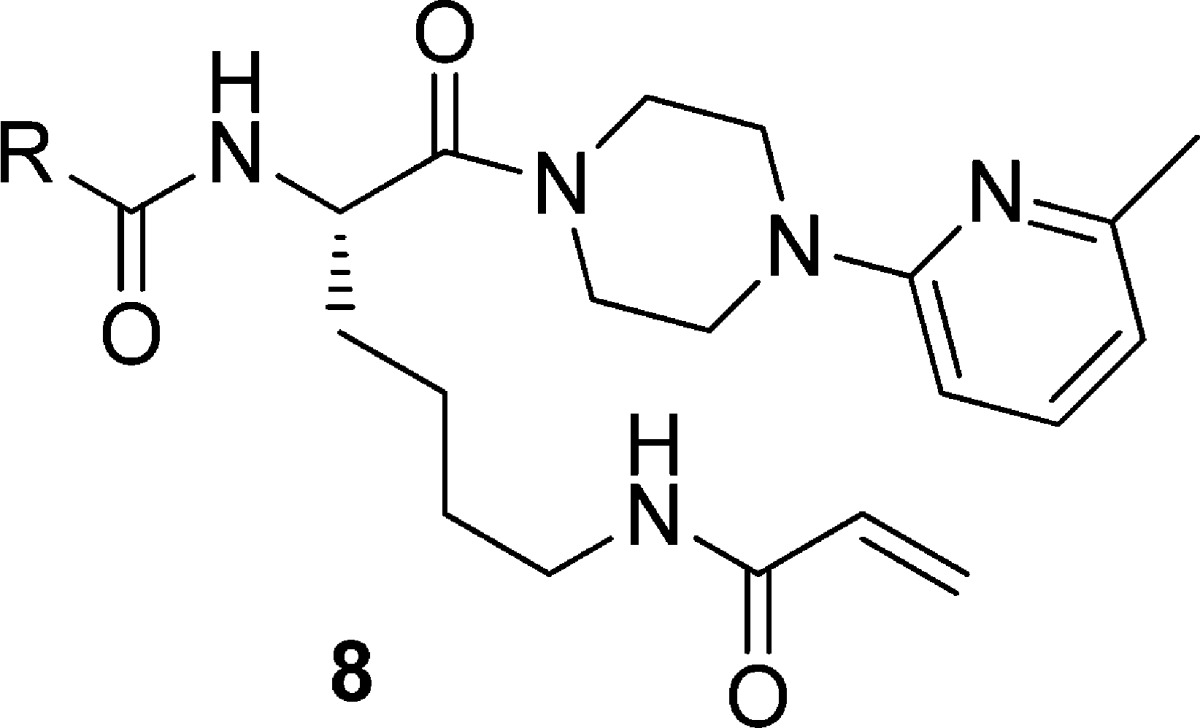
| compd no. | R | PSA (Å2)b | TG2 IC50 ± SD (μM) |
|---|---|---|---|
| 4p | PhCH2O | 104 | 0.055 ± 0.013 |
| 8a | tert-BuO | 104 | 0.85 ± 0.34 |
| 8b | 2-naphthyl | 95 | 1.6 ± 0.021 |
| 8c | CH3 | 95 | 0.19 ± 0.064 |
| 8d | PhCH2 | 95 | 0.014 ± 0.0082 |
| 8e | Ph | 95 | 0.19 ± 0.073 |
Values accompanied by standard deviations were averaged from at least two independent experiments; they were otherwise obtained in a single determination.
PSA calculations were obtained using the Dotmatics mol2image property calculation engine (http://www.dotmatics.com/).
Illustrated in Table 4 are the results of in vitro metabolism profiling of 4l and 8d. In a kinetic solubility assay, both showed good solubility and were stable in both mouse and human plasma with a half-life >24 h. In addition, 4l showed no evidence of conjugation to the prototypical biological nucleophile glutathione (GSH) when tested in vitro over a period of 68 h (data not shown).25 In liver microsomal stability testing, metabolic stability was poor, with a short half-life and rapid intrinsic clearance in both mouse and human. The high rate of metabolism was likely due to the benzyl and piperazinyl moieties, which are susceptible to oxidation at multiple sites.
Table 4. In Vitro ADME Properties of Selected Lysine Amidesa.
| μL/min/mga |
T1/2 (min)a |
|||||||
|---|---|---|---|---|---|---|---|---|
| compd no. | TG2 IC50 (μM) | aq. sol. (mg/mL)a | mLM Clint | hLM Clint | mouse plasma | human plasma | MDCK-WT Papp A–B (nm s–1)a | MDCK-MDR1 EER |
| 8d | 0.014 | 0.87 | NTb | >1386 | >1440 | >1440 | 142 | 118 |
| 4l | 0.06 | 0.06 | >1386 | >1386 | >480 | >480 | 303 | 7.1 |
See the Supporting Information for experimental details.
NT indicates that the compound was not tested.
To be effective agents for HD, it is preferable that compounds possess a high rate of permeability and a low efflux rate. P-glycoprotein (P-gp) is one of the main efflux transporters in brain; the potential for 4l and 8d to be effluxed by P-gp was assessed in an MDCK-MDR1 transfected cell line. The results of this study indicated that both had good permeability but suffered high active efflux via P-gp. Combining this result with the results of microsomal stability testing indicated that these compounds were not suitable candidates for in vivo evaluation in the context of HD. The result of this profiling suggests that they may be better suited as treatments for celiac disease, where BBB permeability would be a disadvantage and 24 h systemic coverage may not be required for efficacy.
In summary, we have developed a series of irreversible TG2 inhibitors based on a known lysine scaffold having improved potency and favorable selectivity against closely related transglutaminase isoforms. The series exhibited low GSH reactivity, improved PSA, and excellent plasma stability but showed a species-dependent loss in potency against mouse TG2. Additionally, compounds exhibited a high rate of oxidative metabolism and high P-gp efflux, signifying that further optimization of the ADME profile will be required to achieve brain exposure in vivo.
Acknowledgments
We are indebted to Ina Sternberger for outstanding technical assistance in conducting transglutaminase biochemical assays. We thank Andreas Ebneth for valuable discussions.
Supporting Information Available
Table of in-house-generated TG2 activity of compounds from Marrano et al.;22 synthetic disclosures comprising schemes and full experimental procedures; and characterization of key compounds, supporting figures, details of the ADME profiling assays, and transglutaminase selectivity profiling data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Greenberg C. S.; Birckbichler R. H. Transglutaminases: Multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991, 5, 3071–3077. [DOI] [PubMed] [Google Scholar]
- Im M.-J.; Riek P.; Graham R. A novel guanine nucleotide-binding protein coupled to the α1-adrenergic receptor. J. Biol. Chem. 1990, 265, 18952–18960. [PubMed] [Google Scholar]
- Lai T.-S.; Slaughter T.; Peoples K.; Hettasch J.; Greenburg C. Regulation of human tissue transglutaminase function by magnesium-nucleotide complexes. J. Biol. Chem. 1998, 273, 1776–1781. [DOI] [PubMed] [Google Scholar]
- Hasegawa G.; Suwa M.; Ichikawa Y.; Ohtsuka T.; Kumagai S.; Kikuchi M.; Sato Y.; Saito Y. A novel function of tissue-type transglutaminase: protein disulfide isomerase. Biochem. J. 2003, 373, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkas D.; Strop P.; Brunger A.; Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLOS Biol. 2007, 5, 2788–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Cerione R. A.; Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2743–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamnaes J.; Pinkas D. M.; Fleckenstein B.; Khosla C.; Sollid L. M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroberardino P. G.; Farrace M.; Viti I.; Pavone F.; Fimia G.; Melino G.; Kodolfo C.; Piacentini M. Tissue transglutaminase contributes to the formation of disulphide bridges in proteins of mitochondrial respiratory complexes. Biochim. Biophys. Acta 2006, 1757, 1357–1365. [DOI] [PubMed] [Google Scholar]
- Molberg O.; McAdam S.; Sollid L. Role of tissue transglutaminase in celiac disease. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 232–240. [DOI] [PubMed] [Google Scholar]
- Mangala L.; Mehta K. In Transglutaminases: The Family of Enzymes with Diverse Functions; Mehta K., Eckert R., Eds.; Karger: Basel, 2005; pp 125–138. [Google Scholar]
- Siegel M.; Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol. Ther. 2007, 115, 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeitner T.; Pinto J.; Krasnikov B.; Horswill M.; Cooper A. Transglutaminases and neurodegeneration. J. Neurochem. 2009, 109, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Sanjuan I.; Bates G. P. The importance of integrating basic and clinical research toward the development of new therapies for Huntington's disease. J. Clin. Invest. 2011, 121, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariello L.; de Cristofaro T.; Zanetti L.; Cuomo T.; Di Maio L.; Campanella G.; Rinaldi S.; Zanetti P.; Di Lauro R.; Varrone S. Transglutaminase activity is related to CAG repeat length in patients with Huntington's disease. Hum. Genet. 1996, 98, 633–5. [DOI] [PubMed] [Google Scholar]
- Mastroberardino P. G.; Iannicola C.; Nardacci R.; Bernassola F.; DeLaurenzi V.; Melino G.; Moreno S.; Pavone F.; Oliverio S.; Fesus L.; Piacentini M. Tissue transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ. 2002, 9, 873–880. [DOI] [PubMed] [Google Scholar]
- Bailey C.; Johnson G. Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J. Neurochem. 2005, 92, 83–92. [DOI] [PubMed] [Google Scholar]
- Chun W.; Lesort M.; Tucholski J.; Faber P. W.; MacDonald M. E.; Ross C. A.; Johnson G. V. W. Tissue transglutaminase selectively modifies proteins associated with truncated mutant huntingtin in intact cells. Neurobiol. Dis. 2001, 8, 391–404. [DOI] [PubMed] [Google Scholar]
- Ruan Q.; Quintanilla R. A.; Johnson G. V. Type 2 transglutaminase differentially modulates striatal cell death in the presence of wild type or mutant huntingtin. J. Neurochem. 2007, 102, 25–36. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Kneynsberg A.; Tucholski J.; Perry G.; van Groen T.; Detloff P. J.; Lesort M. Tissue transglutaminase overexpression does not modify the disease phenotype of the R6/2 mouse model of Huntington's disease. Exp. Neurol. 2012, 237, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keillor J. W.; Chabot N.; Roy I.; Mulani A.; Leogane O.; Pardin C.. Irreversible inhibitors of tissue transglutaminase. In Advances in Enzymology and Related Areas of Molecular Biology; Toone E. J., Ed.; John Wiley & Sons, Inc.: New York, 2011; Vol. 78, pp 415–447. [DOI] [PubMed] [Google Scholar]
- Prime M. E.; Andersen O. A.; Barker J. J.; Brooks M. A.; Cheng R. K. Y.; Toogood-Johnson I.; Courtney S. M.; Brookfield F. A.; Yarnold C. J.; Marston R. W.; Johnson P. D.; Johnsen S. F.; Palfrey J. J.; Vaidya D.; Erfan S.; Ichihara O.; Felicetti B.; Palan S.; Pedret-Dunn A.; Schaertl S.; Sternberger I.; Ebneth A.; Scheel A.; Winkler D.; Toledo-Sherman L.; Beconi M.; Macdonald D.; Muñoz-Sanjuan I.; Dominguez C.; Wityak J. Discovery and SAR of potent and selective covalent inhibitors of transglutaminase 2 for Huntington's disease. J. Med. Chem. 2012, 55, 1021–1046. [DOI] [PubMed] [Google Scholar]
- Prime M. E.; Brookfield F. A.; Courtney S. M.; Gaines S.; Marston R. W.; Ichihara O.; Li M.; Vaidya D.; Williams H.; Pedret-Dunn A.; Reed L.; Schaertl S.; Toledo-Sherman L.; Beconi M.; Macdonald D.; Muñoz-Sanjuan I.; Dominguez C.; Wityak J. Irreversible 4-aminopiperidine transglutaminase 2 inhibitors for Huntington's disease. ACS Med. Chem. Lett. 2012, 3, 731–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrano C.; de Macédo P.; Keillor J. W. Evaluation of novel dipeptide-bound α,β-unsaturated amides and epoxides as irreversible inhibitors of guinea pig liver transglutaminase. Bioorg. Med. Chem. 2001, 9, 1923–1928. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- MacFaul P. A.; Morley A. D.; Crawford J. J. A simple in vitro assay for assessing the reactivity of nitrile containing compounds. Bioorg. Med. Chem. Lett. 2009, 19, 1136–1138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.