

Abstract

Accelerated proliferation of solid tumor and hematologic cancer cells is linked to accelerated transcription of rDNA by the RNA polymerase I (Pol I) enzyme to produce elevated levels of rRNA (rRNA). Indeed, upregulation of Pol I, frequently caused by mutational alterations among tumor suppressors and oncogenes, is required for maintenance of the cancer phenotype and forms the basis for seeking selective inhibitors of Pol I as anticancer therapeutics. 2-(4-Methyl-[1,4]diazepan-1-yl)-5-oxo-5H-7-thia-1,11b-diaza-benzo[c]fluorene-6-carboxylic acid (5-methyl-pyrazin-2-ylmethyl)-amide (CX-5461, 7c) has been identified as the first potent, selective, and orally bioavailable inhibitor of RNA Pol I transcription with in vivo activity in tumor growth efficacy models. The preclinical data support the development of CX-5461 as an anticancer drug with potential for activity in several types of cancer.
Keywords: CX-5461, rRNA, RNA polymerase I, p53
The establishment and maintenance of malignancy depends not only on mutated proto-oncogenes but also on an array of equally essential nonmutated genes that have become deregulated.1,2 Products of these nonmutated genes control processes that enable cancer cells to meet the increased demand in growth and proliferation as well as to deal with the burden of stress imposed by malignant transformation. Such gene products are required to maintain oncogenic signaling and inhibition of nononcogenic signaling, and interference with these proteins can disrupt essential processes upon which cancer cells have become dependent. Surprisingly, such deregulated proteins represent an underexploited group of prime targets for the treatment of cancer.
RNA polymerase I (Pol I) is a prime example of a nononcogene target that can be leveraged for cancer therapeutics. Pol I catalyzes for the synthesis of rRNA (rRNA), which forms the backbone of the ribosome.3−5 The rate of Pol I transcription controls cellular growth and proliferation,6 and under normal conditions, it is tightly regulated. However, during tumorigenesis, the relationship between extracellular signaling and Pol I transcription is deregulated, and cancer cells gain the capacity for excessive production of rRNA and ribosomes necessary to produce proteins for unbridled cell proliferation.6−9 Indeed, upregulation of rRNA is sufficient by itself to maintain the oncogenic phenotype, as siRNA directed at a subunit of Pol I leads to partial inhibition of rRNA synthesis and induction of cell death in cancer cells.10 In addition to the regulation of proliferation, Pol I transcription is also involved in the extensive bidirectional crosstalk with various oncogenes and tumor suppressors.7 One example of such interaction is a relationship of Pol I with tumor suppressor p53. On one hand, activation of p53 is known to repress Pol I transcription through disruption of preinitiation complex formation;11,12 on the other hand, Pol I transcription negatively regulates p53 activation through sequestration of ribosomal proteins in the nucleolus. The inhibition of Pol I transcription was shown to trigger a phenomenon called “nucleolar stress” that leads to the translocation of ribosomal proteins from nucleolus to nucleoplasm, where some of them, for example, rpL5 and rpL11, will bind to MDM2, triggering its dissociation from and thus activation of p53.13 Thus, by sustaining high levels of Pol I transcription, cancer cells maintain nucleolar integrity and hence promote the suppression of p53.
Multiple approved cancer therapeutics have been reported to act through inhibition of rRNA synthesis, but none are known to directly target the Pol I multiprotein enzyme complex.7,14 Hence, the rationale and medical need to justify the search for agents directed at the Pol I complex as a nononcogene target for cancer therapeutics still exist. Therefore, agents that selectively disrupt Pol I transcription are conceptually attractive as anticancer therapeutics.
The goal of the current project was to create a set of molecules that directly, selectively, and potently inhibit Pol I, that are orally bioavailable, and that exert in vivo antitumor activity, from which a clinical candidate could be selected. For this purpose, we developed and deployed a cell-based qRT-PCR assay that measures the differential effects of new chemical entity (NCE) on Pol I- and Pol II-driven transcription, as previously described.15 The in vitro antiproliferative activity of analogues was evaluated using colorectal adenocarcinoma HCT-116 cells and then expanded to additional cell lines. To rapidly screen compounds for oral absorption, a cassette format16,17 was used to dose ICR mice at 25 mg/kg for each compound, and blood was collected at 0.25, 0.5, 1, 2, 4, 6, and 8 h. These efforts led to the rapid discovery of 2-(4-methyl-[1,4]diazepan-1-yl)-5-oxo-5H-7-thia-1,11b-diaza-benzo[c]fluorene-6-carboxylic acid (5-methyl-pyrazin-2-ylmethyl)-amide (CX-5461, 7c), a highly potent, selective, and specific inhibitor of rRNA synthesis that suppresses Pol I transcription at the initiation stage and exhibits antiproliferative activity in vitro and antitumor activity in xenograft models.
The first set of compounds derived from our chemical library, represented by entry 1 (Figure 1 and Table 1), demonstrated reasonable inhibition of Pol I transcription and modest selectivity vis-à-vis a lack of inhibition of Pol II transcription but displayed poor PK characteristics and a lack of oral bioavailability (Table 1). Using scaffold hopping,18 we designed several alterations of the core framework of compound 1 (data not shown) that permit a similar geometric arrangement of the side chains (regions A and C, Figure 1). Among the scaffolds generated from this exercise, the benzothiazole-based scaffold 2 (Figure 1) emerged as a potential lead for a core structure.
Figure 1.

Structures of 1 and 2. Screening lead 2.
Table 1. Selected Analogues SARa.
| IC50 (μM) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | n | X | R1 | R2 | R3 | HCT-116 | rRNA | cMYC | RNA Pol I | selectivity cMYC/rRNA | mouse PO AUC |
| 1 | 0.02 | 0.75 | 5 | ND | 6.6 | NQ | |||||
| 2 | 0.12 | 0.29 | 15.2 | ND | 52.4 | 2586 | |||||
| 1a | 1 | O | H | A | H | 0.05 | 0.07 | 17.5 | 0.91 | 250 | 2398 |
| 2a | 1 | O | F | A | H | 0.08 | 0.16 | 9.05 | 0.63 | 57 | 2261 |
| 3b | 1 | O | F | B | H | 0.59 | 1.25 | 8.17 | 4.41 | 6.5 | 6010 |
| 4a | 1 | O | H | A | F | 0.19 | 0.02 | 3 | ND | 150 | 159 |
| 5c | 1 | O | H | C | H | 0.66 | 0.50 | 2.2 | ND | 4 | 32869 |
| 6c | 1 | NMe | H | C | H | 0.19 | 0.15 | 4.57 | 1.54 | 31 | 90598 |
| 7c | 2 | NMe | H | C | H | 0.15 | 0.22 | 30.93 | 0.88 | 141 | 93373 |
| 8c | 2 | O | H | C | H | >10 | >10 | >10 | >10 | ND | |
| 9c | 2 | Net | H | C | H | 0.17 | 0.13 | 13.37 | 0.64 | 103 | ND |
| 10c | 2 | N-Cyclohexyl | H | C | H | 0.29 | 0.46 | 15.28 | 0.44 | 33 | ND |
AUC(0–24h), ng h/mL; ND, mot determined; and NQ, mot quantifiable.
Compound 2 showed a significant increase in oral absorption while retaining a good in vitro biological profile (Table 1). A systematic study of structure–activity relationships (SARs) was executed for three regions (A–C) of compound 2. The general synthetic route used to prepare these compounds is illustrated in Scheme 1. The synthesis of analogues around compound 2 began with the reaction between substituted 2,6-dichloronicotinic acid chloride 3 and ethyl 2-(5-chlorobenzothiazol-2-yl)acetate 4 in the presence of magnesium chloride to give the desired quinolone.19 Conversion of intermediate 5 to the corresponding substituted amino intermediate 6, followed by formation of an amide group under aluminum chloride-catalyzed reaction conditions, provided quinolone 7. This versatile sequence allowed for the rapid investigation of SARs in all three regions. The key SAR results for select analogues within this series are summarized in Table 1.
Scheme 1. Synthesis of Analogues.

Conditions: (a) MgCl2, Et3N. (b) Amines, NMP, heat. (c) AlCl3, amines, CH2Cl2.
Our initial efforts focused on exploring the effect of electron-withdrawing groups on the core structure (region B). Of note, we found that introduction of fluorine at the R1 position (2a) led to a decrease in RNA Pol I selectivity with similar oral absorption to compound 1a. Furthermore, replacement of hydrogen by fluorine at R3 (4a) resulted in a dramatic loss as compared to compound 2a in oral absorption, although the cellular activity profile was retained.
We next examined modifications to region C. The change of the position of the basic nitrogen from C3 (compound 2) to C2 (compound 1a) on the side chain of region C led to an increase in antiproliferative activity as determined by inhibition of HCT-116 cancer cells and an increase of potency in the inhibition of rRNA synthesis.
This compound also demonstrated higher selectivity for RNA Pol I (rRNA synthesis) versus Pol II (c-myc mRNA synthesis). However, oral absorption was similar for both compounds (2 and 1a). The addition of rigidity to region C (3b) led to a dramatic decrease in cellular and Pol I enzyme activities (IC50 of 0.59 and 4.41 μM, respectively). The selectivity between RNA Pol I and Pol II was also substantially lower (6.5-fold). However, compound 3b demonstrated an oral absorption 2-fold greater than the oral absorption of compound 2a. A breakthrough in increasing AUC was realized when the basic methylpyrrolidine in compound 1a was replaced with methylpyrazine (5c). Although the cellular activity of compound 5c was moderate (IC50 = 0.66 μM), the inhibition of Pol I and Pol II enzymes was completely lost. To regain potency for Pol I enzyme inhibition, we examined the SAR of the morpholino moiety (region A) of compound 5c. A detailed survey of substitutions in region A led to the identification of a few preferred substituents at region A, including 1-methylpiperazine (6c) and 1-methyl-1,4-diazepane (7c). Compound 6c increased oral exposure in mice 3-fold (6c vs 5c) with good overall cellular and enzyme activity profiles. Compound 7c also exhibited higher oral exposure and better selectivity for Pol I as compared to compounds 5c and 6c. Compound 7c presented similar activity to compound 6c in inhibiting cell growth (IC50 = 0.15 μM). The removal of basic center in region A (8c) led to complete loss of biological activity. Increasing the size of N-substituents on piperazine ring (9c and 10c) did not alter the overall cellular or enzymatic activities.
On the basis of target selectivity, in vitro antiproliferative activity, and preliminary in vivo PK properties, compound 7c was selected for further in vitro and in vivo testing. Treatment of biphenotypic B myelomonocytic leukemia cell line MV 4;11 and large cell immunoblastic lymphoma cell line SR with compound 7c resulted in the suppression of Pol I transcription in both cell lines with EC50 values of 95 and 135 nM, respectively. Furthermore, compound 7c demonstrated approximately 200-fold selectivity against Pol I relative to Pol II, while exerting no inhibition of DNA replication or protein translation at concentrations as high as 10 μM. Compound 7c also demonstrated robust cell growth inhibition in both cell lines with IC50 values of 11 and 13 nM, respectively. Compound 7c caused a significant activation of p53 as well as its transcriptional target p21 in MV 4;11 and SR cells at 1 μM concentration (Table 2).
Table 2. Effect of 7c on Activation of p53 and p21a.
| cell line | p53 protein levels | p21 protein levels |
|---|---|---|
| MV 4;11 | 6.7 | 5.4 |
| SR | 4.1 | 3.2 |
Activation of p53 and p21. Cells were treated for 6 h with 7c. The induction of p53 and p21 was measured by Western hybridization.
COMET assay20 ruled out the possibility that the activation of p53 observed in both cell lines is due to DNA damage, a process also known to activate p53,21 but rather to Pol I inhibition. As Figure 2 in the Supporting Information shows, 7c had no noticeable effect on DNA integrity relative to the positive control actinomycin, a known DNA-damaging agent, at similar concentrations (100 nM). At 10 μM CX-5461, the COMET formation was more evident, but this concentration of 7c is approximately 100-fold the antiproliferative EC50, and the effect is likely to be secondary to the induction of cell death and the associated DNA fragmentation.
Figure 2.
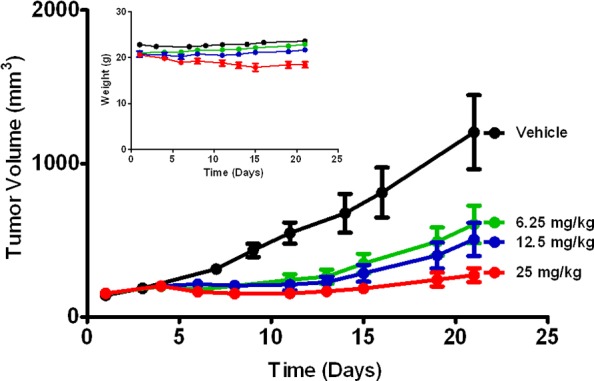
Tumor growth efficacy of 7c in MV 4;11 xenograft as measured by tumor volume. Nude mice were administered 6.25, 12, or 25 mg/kg dose ip once a day on a 5-on, 2-off schedule over a period of 21 days. Also represented is the 21 days animal body weight. Error bars denote the standard error of the mean (SEM).
In addition, 7c was shown to be neither a strong DNA intercalator nor minor groove binding drug at a concentration as high as 50 μM (Figure 3 and the Supporting Information). Taken together, these mechanistic assays demonstrated that the activation of p53 is due to Pol I inhibition.
Figure 3.
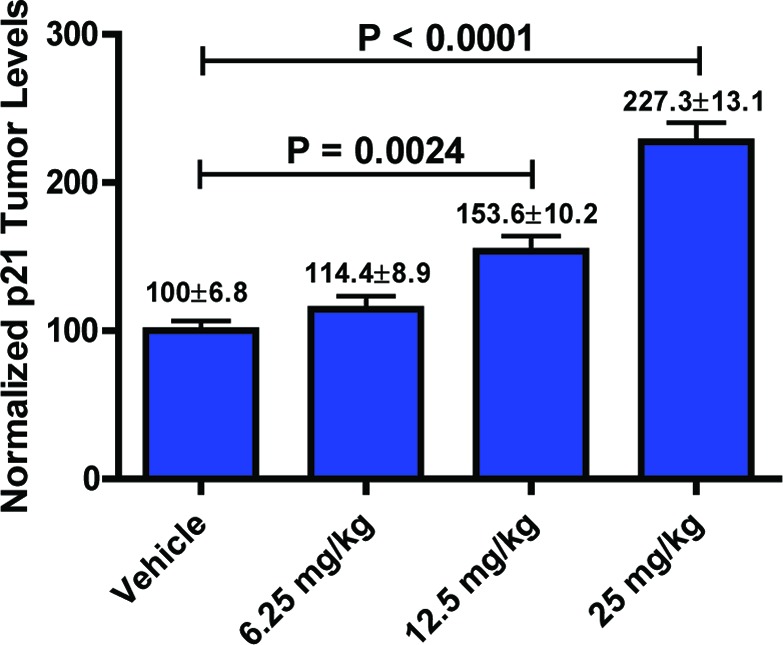
PD biomarker study. Nude mice bearing MV 4;11 xenografts were administered single 6.25, 12, or 25 mg/kg ip dose of 7c. Eight hours later, animals were sacrificed, and intratumoral levels of p21 were determined by Western hybridization. Error bars denote the standard error of the mean (SEM).
The PK profile of 7c was evaluated in four preclinical species (mouse, rat, dog, and monkey). Favorable half-lives (T1/2) were observed across species with this compound. Clearance was low to moderate, and the volume of distribution (Vd) was larger than total body water in dog and monkey, indicating extensive extravascular distribution. The measured oral bioavailability ranged from 24% in mice to 45% in Cynomolgus monkey (Table 3).
Table 3. Preclinical PK Profile of 7ca.
| iv dosing |
oral dosing |
||||
|---|---|---|---|---|---|
| species | Cls | Vd | T1/2 | AUC | % F |
| mouse | 0.03 | 0.24 | 6.3 | 93373 | 24 |
| rat | 0.06 | 0.08 | 5.9 | 92057 | 29 |
| dog | 1.6 | 21 | 8.9 | 3890 | 38 |
| monkey | 1.8 | 33 | 12.8 | 2597 | 45 |
Units: Cls, L/h/kg; Vd, L/kg; T1/2, h; and AUC(0–24 h), ng h/mL. Vehicle, 25 mM NaH2PO4, pH 5.95; data are reported as an average of three animals.
Previously, we reported that 7c, administered orally, had good in vivo antitumor activities against human solid tumors in murine xenograft models.15 To expand our options of route of administration, we assessed the antitumor activity of 7c, administered ip, in a murine MV 4;11 xenograft model. Once daily, ip administration of 7c resulted in a dose-dependent inhibition of tumor growth (Figure 2). At 25 mg/kg, the compound achieved a significant delay in tumor growth (TGI of 84%). All tested doses were well-tolerated as observed by the absence of significant changes in animal body weights (Figure 2).
To test if antitumor activity of 7c correlated with induction of p53 in tumor, we performed a pharmacodynamics (PD) biomarker study. Mice carrying MV 4;11 xenograft received a single dose of either 6.25, 12, or 25 mg/kg. Six hours after dose administration, the levels of p53 transcriptional target, p21, were determined in tumors. Compound 7c was found to induce p21 in MV 4;11 xenografts in dose-dependent manner (Figure 3).
As part of the pharmaceutical assessment, compound 7c was subjected to a series of preclinical safety assays. When tested against the major CYP450 drug-metabolizing enzymes, the IC50 values were greater than 10 μM, indicating that 7c would be unlikely to influence the metabolism of other drugs. In an automated hERG patch-clamp assay, 10 μM 7a showed no significant inhibitory effect on conduction, indicating that 7c would be unlikely to influence cardiac function. Furthermore, compound 7c was negative in tests for mutogenicity (Ames). When tested for potential off-target effects against a large panel of receptors, ion channels, transporters, kinases, and proteases, 7c was found to have a favorable profile.
In summary, we report the discovery of 7c, the first known direct and selective inhibitor of Pol I transcription. Compound 7c displays high potency in both mechanistic and antiproliferative cellular assays, is orally bioavailable, and exhibits potent antitumor activity in solid and hematological xenograft models. Inhibition of rRNA synthesis through Pol I modulation is expected to have a beneficial effect on cancer therapy. Compound 7c is undergoing further studies to identify the most appropriate cancer indications for the clinical evaluation of this innovative agent.
Supporting Information Available
Biological assays and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Solimini N. L.; Luo J.; Elledge S. J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 1306986–988. [DOI] [PubMed] [Google Scholar]
- Luo J.; Solimini N. L.; Elledge S. J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 1365823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummt I. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003, 17141691–1702. [DOI] [PubMed] [Google Scholar]
- Moss T. At the crossroads of growth control; making ribosomal RNA. Curr. Opin. Genet. Dev. 2004, 142210–217. [DOI] [PubMed] [Google Scholar]
- Russell J.; Zomerdijk J. C. The RNA polymerase I transcription machinery. Biochem. Soc. Symp. 2006, 73, 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D.; Pandolfi P. P. Does the ribosome translate cancer?. Nat. Rev. Cancer 2003, 33179–192. [DOI] [PubMed] [Google Scholar]
- Drygin D.; Rice W. G.; Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [DOI] [PubMed] [Google Scholar]
- White R. J. RNA polymerases I and III, growth control and cancer. Nat. Rev. Mol. Cell Biol. 2005, 6169–78. [DOI] [PubMed] [Google Scholar]
- White R. J. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008, 2412622–629. [DOI] [PubMed] [Google Scholar]
- Bywater M.; Poortinga G.; Cullinane G.; Stanley K.; Walker R.; Drygin D.; Anderes K.; Johnstone R.; McArthur G.; Hannan R. In RNA Polymerase I: A Novel Target in the Treatment of MYC-Driven Malignancy, AACR Special Conference on Protein and Cancer San Diego, CA, 2010; San Diego, CA, 2010.
- Budde A.; Grummt I. p53 represses ribosomal gene transcription. Oncogene 1999, 1841119–1124. [DOI] [PubMed] [Google Scholar]
- Zhai W.; Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 2000, 20165930–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisenroth C.; Zhang Y. Ribosome biogenesis surveillance: Probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 2010, 29304253–4260. [DOI] [PubMed] [Google Scholar]
- Burger K.; Muhl B.; Harasim T.; Rohrmoser M.; Malamoussi A.; Orban M.; Kellner M.; Gruber-Eber A.; Kremmer E.; Holzel M.; Eick D. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 2851612416–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drygin D.; Lin A.; Bliesath J.; Ho C. B.; S.; Proffitt C.; Omori M.; Haddach M.; Schwaebe M.; Siddiqui-Jain A.; Streiner N.; Quin J. E.; Sanij E.; Bywater M. J.; Hannan R. D.; Ryckman D.; Anderes K.; Rice W. G. RNA Polymerse I Targeting with an Oral Small Molecule CX-5461 Inhibits Ribosomal RNA Synthesis and Solid Tumor Growth. Cancer Res. 2011, 71, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Smith N. F.; Raynaud F. I.; Workman P. The application of cassette dosing for pharmacokinetic screening in small-molecule cancer drug discovery. Mol. Cancer Ther. 2007, 62428–440. [DOI] [PubMed] [Google Scholar]
- Smith N. F.; Hayes A.; Nutley B. P.; Raynaud F. I.; Workman P. Evaluation of the cassette dosing approach for assessing the pharmacokinetics of geldanamycin analogues in mice. Cancer Chemother. Pharmacol. 2004, 546475–486. [DOI] [PubMed] [Google Scholar]
- Brown N.; Jacoby E. On scaffolds and hopping in medicinal chemistry. Mini Rev. Med. Chem. 2006, 6111217–1229. [DOI] [PubMed] [Google Scholar]
- Chua P. C.; Nagasawa J. Y.; Nagasawa J.; Pierre F.; Schaebe M. K.; Vialettes A.; Whitten J. P. A novel and efficient synthesis of 3-carboxy-4-oxo-1,8-naphthyridines using magnesium chloride. Tetrahedron Lett. 2008, 49, 4437–4442. [Google Scholar]
- Miyamae Y.; Zaizen K.; Ohara K.; Mine Y.; Sasaki Y. F. Detection of DNA lesions induced by chemical mutagens by the single cell electrophoresis (Comet) assay. 1. Relationship between the onset of DNA damage and the characteristics of mutagens. Mutat. Res. 1998, 4153229–235. [DOI] [PubMed] [Google Scholar]
- Gotz C.; Montenarh M. p53: DNA damage, DNA repair, and apoptosis. Rev. Physiol. Biochem. Pharmacol. 1996, 127, 65–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.