

Abstract

Extensive structure–activity relationship studies of a series derived from atropisomer 1, a previously described chiral benzodiazepine sulfonamide series, led to a potent, brain penetrant and selective compound with excellent preclinical pharmacokinetic across species. We also describe the utilization of a high throughput mouse pharmacodynamic assay which allowed for expedient assessment of pharmacokinetic and brain distribution.
Keywords: Bombesin receptor subtype-3, BRS-3, agonist, obesity, benzodiazepine sulfonamide, atropisomer, MK-7725
Approximately 30%–40% of marketed drugs directly or indirectly target 50 of the 400 nonolfactory G protein-coupled receptors, known as GPCRs, or as 7-TMRs (seven transmembrane receptors). The key feature of the GPCR family is the seven membrane-spanning domains or transmembrane helices that interact with signaling molecules outside the cell to activate signal transduction pathways inside the cell resulting in cellular responses. There are more than 250 “known” GPCRs, for which the endogenous ligands have been identified, while for the rest the endogenous ligands have not been identified.1 These are collectively known as “orphan” receptors. Our laboratory had significant interest in the orphan GPCR bombesin receptor subtype-3 (BRS-3 or BB3) as a possible drug target to modulate energy homeostasis based upon rodent genetic validation studies.2,3 BRS-3 is expressed primarily in the central nervous system (CNS), thus making a brain penetrant compound a necessity.4−10
In this letter, we describe the design and synthesis of a second generation benzodiazepine BRS-3 agonist derived from our previously described compound 1 (shown in Figure 1).11 A major hurdle for development of compound 1 would be the poor preclinical pharmacokinetics (PK) that we sought to optimize. To assist us in this endeavor, we describe access to a mouse pharmacodynamic (PD) model which allowed for rapid assessment of multiple compounds. This allowed for inference of in vivo absorption and distribution to the brain where the BRS-3 receptors reside.12 In addition to optimization of brain penetration, we sought to improve upon the pregnane X receptor (PXR) activation which plagued compound 1 (EC50 = 677 nM, 42% @ 10 μM). Activation of the nuclear hormone receptor PXR leads to upregulation of CYP enzymes, which would make drug–drug interactions for this class of compounds a major concern for subsequent development.
Figure 1.

Benzodiazepine lead.
Since the BRS-3 receptor resides in the brain, distribution into the brain is necessary for any candidate. Brain exposure studies can provide some information, but they cannot account for tissue binding. To facilitate the structure–activity relationship (SAR) advancement, we needed a mechanism-based readout for rapid assessment of BRS-3 receptor activation after a compound was dosed. Our lab discovered that treatment of mice with a BRS-3 agonist causes a core body temperature increase, likely as a surrogate of increasing metabolic rate. This effect is absent in BRS-3 knockout mice, suggesting the observed temperature increase is mediated through the BRS-3 receptor.
As illustrated in Figure 2, vehicle treated mice experienced a transient core temperature increase corresponding to activity change during handling. The core temperature stabilized 1–2 h post dosing. When mice were treated with compound 2, a previously described BRS-3 agonist with excellent functional potency in the mouse (mEC50 = 2.5 nM),6 a dose-dependent increase in body temperature and duration was observed. To facilitate comparison among compounds, area under the curve calculations (AUC) at either 1–2 h or 1–6 h were computed. The time period between 0 and 1 h post dose was ignored because the core temperature change in this period is largely related to animal handling and not to the compound. The core temperature can be readily monitored by an implanted minisensor, making this assay (FAST PD: fasted temperature PD assay in DIO mice) highly reproducible and allowed for the measurement of multiple compounds per week.
Figure 2.
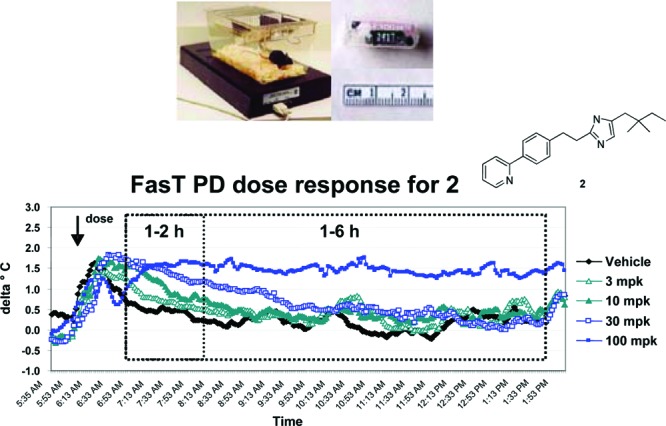
FAST PD dose response to a previously reported BRS-3 agonist.7
With a high throughput PD assay now in hand, our objectives for a diazepine structurally related to 1 were to address the short half-life and PXR activation associated with lead compound 1. We commenced our SAR studies by expanding the substitution at the 2-position of the benzodiazepine core. We devised a synthesis which would allow for facile derivatization of the 2-position of the benzodiazepine while also allowing for a penultimate chiral separation to occur on each derivative which is planar chiral, as previously described in detail.11,13
The synthesis of the 2-substituted diazepine sulfonamide analogues was accomplished through condensation of 3-chlorobenzene-1,2-diamine (3) and 2-chloro-6-(trifluoromethyl) nicotinic acid (4) under thermal conditions, which provided amide 5. Amide 5 was reduced by borane to yield the respective diazepine 6. Subsequent sulfonylation gave rise to the desired diazepine sulfonamide 7 (Scheme 1).
Scheme 1. Synthesis of 2-Substituted Diazepine Sulfonamide Analogues.

Conditions: (a) 2-butoxyethanol, 150°C, 12 h; (b) BH3·THF; (c) DCM, pyridine.
Suzuki coupling chemistry was employed to introduce various heterocycles at the 2-position (Scheme 2).14 Most of the analogues prepared displayed potent in vitro activity. For example, a 3-pyridyl substituent, 9a, displayed excellent binding (hBRS-3, IC50 1.7 nM) and functional activity (hBRS-3, EC50 42 nM) while a 5-member heterocycle such as pyrazole 9b is also functionally active (hBRS-3, EC50 63 nM) on the BRS-3 receptor. This would seem to indicate a tolerance for substitution of heterocycles at the 2-position. The piperidine derivative 9c is a potent binder (hBRS-3, IC50 1.4 nM) but has reduced functional activity (hBRS-3, EC50 114 nM) in the in vitro assay. Encouragingly, compound 9c showed reduced PXR activation (6.6 μM, 37%) which is a key objective for this lead class.
Scheme 2. Introduction of Various Heterocycles at the 2-Position.

Conditions: (a) Pd(OAc)2, S-Phos, R1B(OH)2, K3PO4, 10:1 toluene/H2O, 150 °C; (b) chiral separation (Chiralpak OD, EtOH/hexanes).
De novo synthesis of these substituted heterocycles started with the 2-CN intermediate, obtained by cyanation of the chloro precursor 8.15 Formation of the amide oxime using hydroxyl amine, subsequent acylation, and dehydration now provided access to ring systems previously unavailable through Suzuki coupling.16 The parent 1,2,4-oxadiazole 9e was synthesized (Scheme 3) and displayed excellent binding (hBRS-3, IC50 1.1 nM) and functional activity (hEC50 = 40 nM) on the human BRS-3 receptor as well as good mouse functional activity (mBRS-3, EC50 57 nM). In addition, 9e displayed moderate PD efficacy at the 3 mpk dose with an improvement in PXR activation (11.8 μM, 53%).
Scheme 3. De novo Oxadiazole Synthesis.

Conditions: (a) Pd(OAc)2, S-Phos, Zn(CN)2, 99:1 DMF:H2O. (b) NH2- OH. (c) R1COCl, pyridine, DMAP or R1COOH, PyBOP, DMF then TBAF, THF (d) Chiral separation (Chiralpak OD, EtOH/hexane).17
More consistent improvement in lowering PXR activation was achieved when a polar substituent was introduced (Table 1), also with concomitant improvement in solubility. Among the primary alcohol 9f,two secondary alcohols (9g and 9 h), and the tertiary alcohol 9i, compound 9i had displayed excellent in vitro potency (hBRS-3, IC50 0.9 nM), good PD efficacy, and reduced PXR activation (>10 μM, 13%).
Table 1. Various Heterocyclic Substituted Diazepenes' Human Binding/Functional Activity and Mouse Functional BRS-3 Activity.
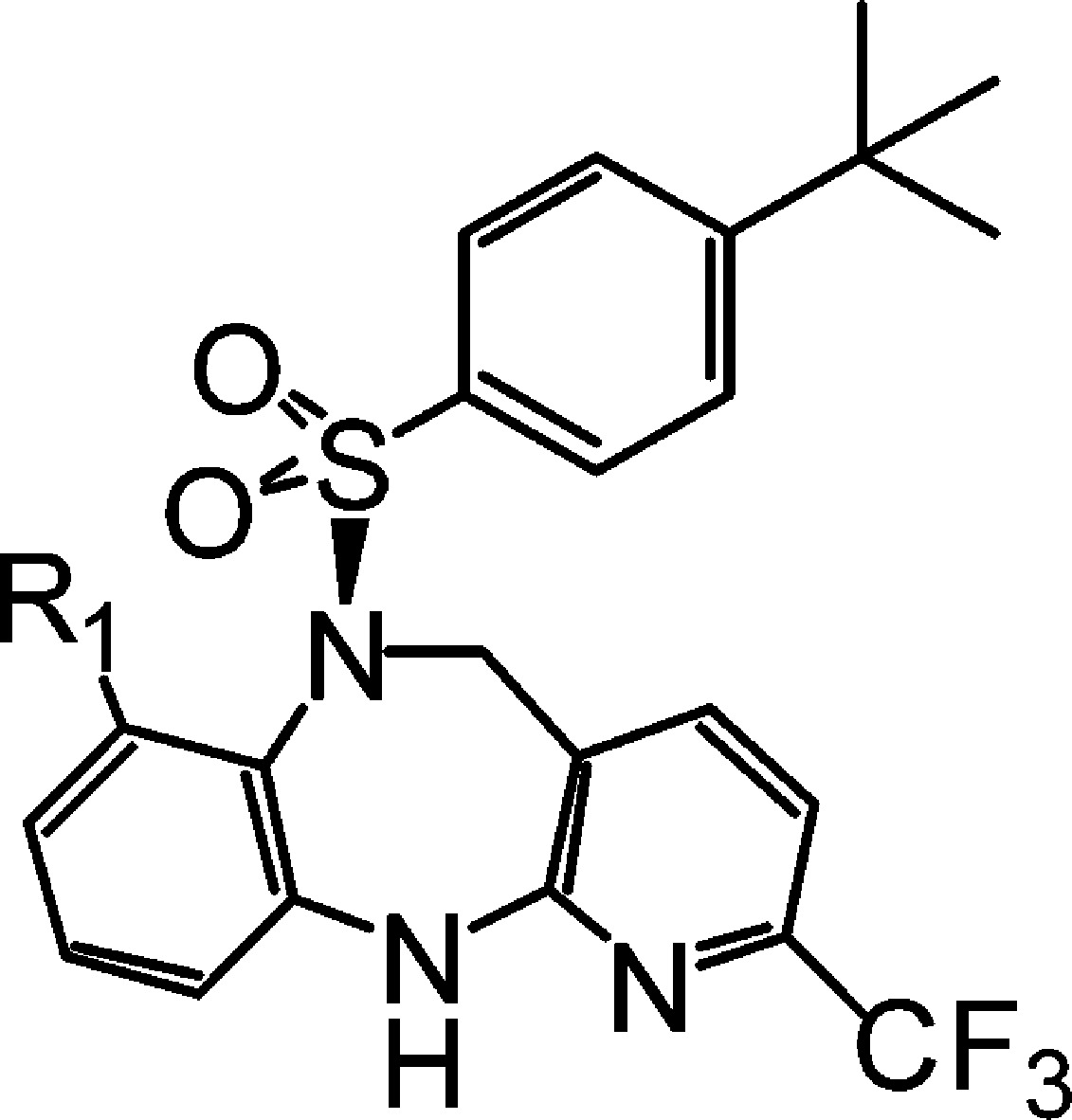
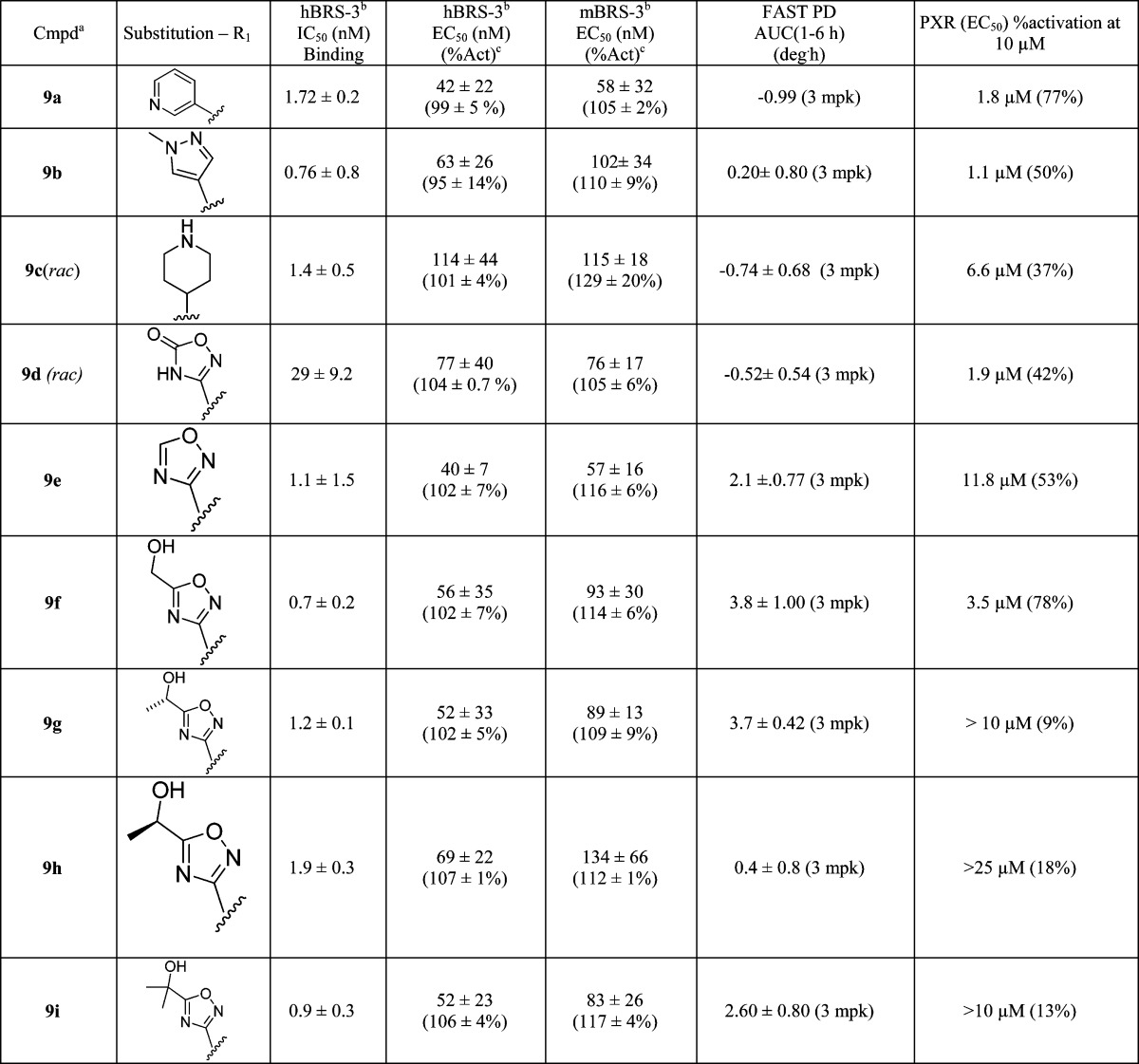
Enantiomerically pure compounds unless otherwise noted.
Data expressed as mean ± SD (n ≥ 2 independent experiments).
%Act represents the maximum activation of tested compound relative to that of the known BRS-3 agonist dY peptide compound.
It is worth noting that the trend in PD efficacy is somewhat difficult to interpret. To better discern the observed data, rat pharmacokinetic studies were performed on selected compounds (Table 2). On the basis of the PD, PK, and binding or functional data, we could surmise whether the dominant factor for PD efficacy is pharmacokinetic properties or brain penetration, as opposed to solely in vitro potency. Compound 9g showed excellent activity in the FAST assay (3.7 deg·h) but suffered from high clearance (86) and low exposure (0.34). Compounds 9f and 9i seemed to both have good FAST activity and T1/2. Both compounds were of interest, but the liability of in vivo oxidation of the primary alcohol began to dissuade us from pursuing compounds such as 9f any further. Compound 9i was the subject of further SAR studies to improve its preclinical PK profile and bioavailability.
Table 2. Rat Pharmacokinetic Dataa of Selected Compoundsb.
| cmpd | Clp ((mL/min)/kg) | T1/2 (h) | PO AUCN (μM·h·kg/mg) | Foral (%) | FAST (deg·h) |
|---|---|---|---|---|---|
| 9f | 22 | 1.7 | 0.39 | 29 | 2.6 |
| 9g | 86 | 2.1 | 0.34 | 14 | 3.7 |
| 9h | 33 | 1.8 | 0.90 | 11 | 0.4 |
| 9i | 44 | 1.9 | 0.65 | 14 | 2.6 |
Sprague–Dawley (SD) rat was used.
Plasma clearance (Clp) and half-life (T1/2) calculated following 0.5 mg/kg IV dose. Normalized oral exposure (PO AUCN) and oral bioavailability (Foral) calculated following 1.0 mg/kg PO dose.
Additional modification of 9i was carried out on the sulfonamide phenyl substituent (Table 3). Replacement of the tert-butyl with an isopropyl group led to compound 12, which showed good overall potency and PD efficacy. Not surprisingly, no improvement in PK was observed (Clp = 49, T1/2 = 1.6 h, F% = 8), suggesting the improvement in PD is likely a result of improved brain penetration. Measured rat plasma protein binding in these particular instances did not provide an adequate enough window to infer a higher level of free fraction based on measured values to account for the PD efficacy difference.18 Among the fluorinated analogues 13, 14, and 15, the trifluoromethoxy analogue 14 displayed a robust efficacy in vivo PD, as well as a superior rat PK to analogue 13 as shown in Table 4. These factors led us to further profile the compound in higher species pharmacokinetic studies.
Table 3. Sulfonamide Variation.
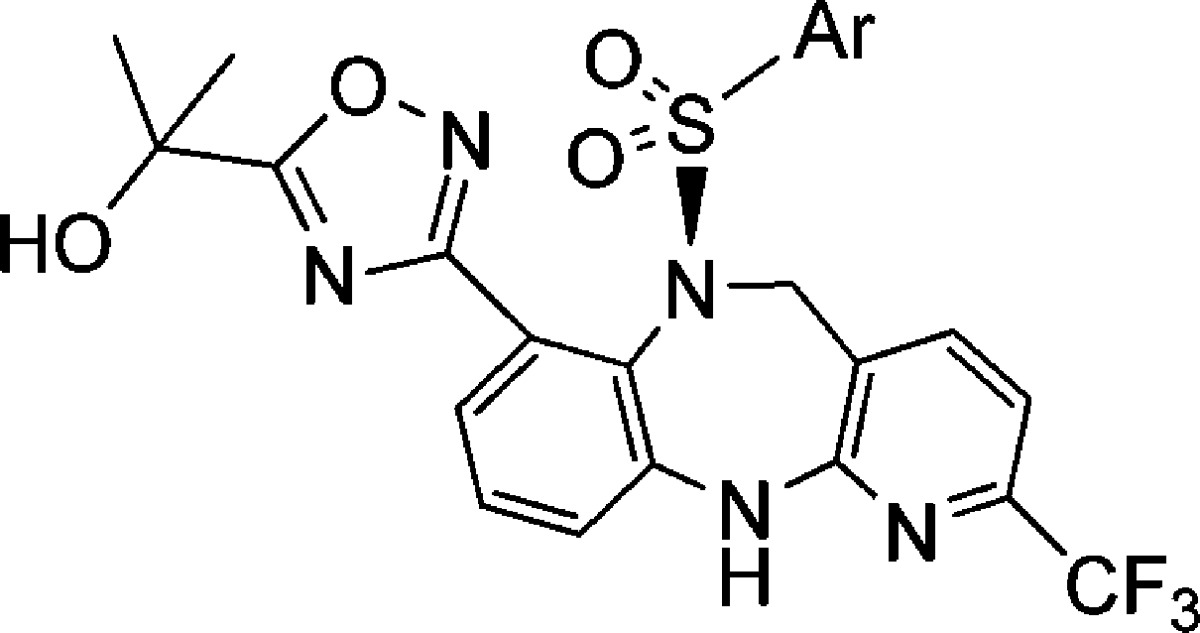

Table 4. Pharmacokinetic Dataa of Compounds 13 and 14.
| species | Clp ((mL/min)/kg) | T1/2 (h) | PO AUCN (μM·h·kg/mg) | Foral (%) |
|---|---|---|---|---|
| rat-(13) | 33 | 2.5 | 1.05 | 100 |
| rat-(14) | 16 | 11 | 1.60 | 93 |
| dog-(14) | 2.9 | 18 | 10.0 | 100 |
| rhesus-(14) | 1.3 | 33 | 12.0 | 58 |
Plasma clearance (Clp) and half-life (T1/2) calculated following 0.5 mg/kg IV dose. Normalized oral exposure (PO AUCN) and oral bioavailability (Foral) calculated following 1.0 mg/kg PO dose.
Compound 14 showed excellent PK across several preclinical species and was devoid of PXR activation that plagued our lead compound 1. Compound 14 was assessed for acute efficacy in wild-type (WT) and BRS-3 knockout (KO) mice. In WT mice, the compound caused a food intake reduction and body weight loss in a dose-dependent manner when dosed at 10, 30, and 100 mpk (see Figure 3). In KO mice, no effect was observed at 100 mpk PO.19 A 12-day chronic study was initiated in DIO rats at 15 and 50 mpk BID, and robust weight loss was observed at the end of the study with no evidence of tachyphylaxis. The body weight loss was shown to be a combination of reduction in food intake and increase in metabolic rate (data not shown). On the basis of the data generated, compound 14 was accepted for further development as a preclinical candidate and designated MK-7725.
Figure 3.

Effect of 14 on body weight in DIO rats (15 and 50 mpk BID).
A follow up weight loss study was also carried out in obese female dogs, and ∼4% weight loss was observed at both the 5 and 15 mpk dose after 7 days of treatment with MK-7725 (Figure 4).20
Figure 4.

Effect of MK-7725 on body weight in female obese dogs (5 and 15 mpk).
In summary, a highly efficient PD assay was developed to accelerate triaging of novel compounds synthesized, and SAR effort of the diazepine class resulted in the identification of compounds with improved pharmacokinetic properties and reduced PXR activation relative to the initial lead. On the basis of the overall profile, compound 14 (designated as MK-7725) was selected as a structurally distinct backup candidate to previously described MK-50469 for clinical evaluation as a treatment for obesity.
Acknowledgments
The author wishes to thank the department of Laboratory Animal Resources for their assistance in animal dosing and sampling as well as Dr. Andrea Peier for helpful suggestions.
Supporting Information Available
Synthetic procedures and analytical data of selected BRS-3 agonists, and conditions for all the biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Blakeney J. S.; Reid R. C.; Le G. T.; Fairlie D. P. Nonpeptidic Ligands for Peptide-Activated G Protein-Coupled Receptors. Chem. Rev. 2007, 107, 2960. [DOI] [PubMed] [Google Scholar]
- Ohki-Hamazaki H.; Watase K.; Yamamoto K.; Ogura H.; Yamano M.; Yamada K.; Maeno H.; Imaki J.; Kikuyama S.; Wada E.; Wada K. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature 1997, 390, 165–169. [DOI] [PubMed] [Google Scholar]
- b Ladenheim E. E.; Hamilton N. L.; Behles R. R.; Bi S.; Hampton L. L.; Battey J. F.; Moran T. H. Factors Contributing to Obesity in Bombesin Receptor Subtype-3-Deficient Mice. Endocrinology 2008, 149, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber D.; Berger C.; Eickelmann P.; Antel J.; Kessler H. Design of Selective Peptidomimetic Agonists for the Human Orphan Receptor BRS-3. J. Med. Chem. 2003, 46, 1918–1930. [DOI] [PubMed] [Google Scholar]
- Carlton D. L.; Collin-Smith L. J.; Daniels A. J.; Deaton D. N.; Goetz A. S.; Laudeman C. P.; Littleton T. R.; Musso D. L.; Ott Morgan R. J.; Szewczyk J. R.; Zhang C. Discovery of Small Molecule Agonists for the Bombesin Receptor Subtype 3 (BRS-3) Based on an Omeprazole Lead. Bioorg. Med. Chem. Lett. 2008, 18, 5451–5455. [DOI] [PubMed] [Google Scholar]
- He S.; Dobbelaar P. H.; Liu J.; Jian T.; Sebhat I. K.; Lin L. S.; Goodman A.; Guo C.; Guzzo P. R.; Hadden M.; Henderson A. J.; Ruenz M.; Sargent B.; Yet L.; Kelly T. M.; Palyha O.; Kan Y.; Pan J.; Chen H.; Marsh D.; Shearman L. P.; Strack A. M.; Metzger J. M.; Feighner S. D.; Tan C.; Howard A. D.; Tamvakopoulos C.; Peng Q.; Guan X.-M.; Reitman M. L.; Patchett A. A.; Wyvratt M. J.; Nargund R. P. Discovery of Substituted Biphenyl Imidazoles as Potent Bioavailable Bombesin Receptor Subtype-3 Agonists. Bioorg. Med. Chem. Lett. 2010, 20, 1913–1917. [DOI] [PubMed] [Google Scholar]
- Liu J.; He S.; Jian T.; Dobbelaar P. H.; Sebhat I. K.; Lin L. S.; Goodman A.; Guo C.; Guzzo P. R.; Hadden M.; Henderson A. J.; Pattamana K.; Ruenz M.; Sargent B.; Swenson B.; Yet L.; Tamvakopoulos C.; Peng Q.; Pan J.; Kan Y.; Palyha O.; Kelly T. M.; Guan X.-M.; Howard A. D.; Marsh D.; Metzger J.; Reitman M. L.; Wyvratt M. J.; Nargund R. P. Synthesis and SAR of Derivatives Based on 2-Biarylethylimidazole as Bombesin Receptor Subtype-3 (BRS-3) Agonists for the Treatment of Obesity. Bioorg. Med. Chem. Lett. 2010, 20, 2074–2077. [DOI] [PubMed] [Google Scholar]
- Hadden M.; Goodman A.; Guo C.; Guzzo P. R.; Henderson A. J.; Pattamana K.; Ruenz M.; Sargent B.; Swenson B.; Yet L.; Liu J.; He S.; Sebhat I. K.; Lin L. S.; Tamvakopoulos C.; Peng Q.; Kan Y.; Palyha O.; Kelly T. M.; Guan X.-M.; Metzger J.; Reitman M. L.; Nargund R. P. Synthesis and SAR of Heterocyclic Carboxylic Acid Isosteres Based on 2-Biarylethylimidazole as Bombesin Receptor Subtype-3 (BRS-3) Agonists for the Treatment of Obesity. Bioorg. Med. Chem. Lett. 2010, 20, 2912–2915. [DOI] [PubMed] [Google Scholar]
- Sebhat I. K.; Franklin C.; Lo M. M.; Chen D.; Jewell J. P.; Miller R.; Pang J.; Palyha O.; Kan Y.; Kelly T. M.; Guan X.-M.; Marsh D. J.; Kosinski J. A.; Metzger J. M.; Lyons K.; Dragovic J.; Guzzo P. R.; Reitman M. L.; Nargund R. P.; Wyvratt M. J.; Lin L. S. Discovery of (2S)-1,1,1-Trifluoro-2-[4-(1H-pyrazol-1-yl)phenyl]-3-(4-{[1-(trifluoromethyl)cyclopropyl]methyl}-1H-imidazol-2-yl)propan-2-ol (MK-5046), a Potent, Selective Bombesin Receptor Subtype-3 (BRS-3) Agonist for the Treatment of Obesity. ACS Med. Chem. Lett. 2011, 2, 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M. M.-C.; Chobanian H. R.; Palyha O.; Kan Y.; Kelly T. M.; Guan X.-M.; Reitman M. L.; Dragovic J.; Lyons K. A.; Nargund R. P.; Lin L. S. Pyridinesulfonylureas and Pyridinesulfonylamides as Selective Bombesin Receptor Subtype-3 (BRS-3) Agonists. Bioorg. Med. Chem. Lett. 2011, 21, 2040–2043. [DOI] [PubMed] [Google Scholar]
- Liu P.; Lanza T. J.; Chioda M.; Jones C.; Chobanian H. R.; Guo Y.; Chang L.; Kelly T. M.; Kan Y.; Palya O.; Guan X.; Marsh D. J.; Metzger J. M.; Raustad K.; Wang S.; Strack. A. M.; Miller R.; Pang J.; Lyons K.; Dragovic J.; Ning J. G.; Schafer W. A.; Welch C. J.; Gong X.; Gao Y.; Hornak V.; Ball R. G.; Tsou N.; Reitman M. L.; Wyvratt M. J.; Nargund R. P.; Lin L. S. Discovery of Benzodiazepine Sulfonamide-Based Bombesin Receptor Subtype 3 Agonist for the Treatment of Obesity. ACS Med. Chem. Lett. 2011, 2, 933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X.-M.; Chen H.; Dobbelaar P. H.; Dong Y.; Fong T. M.; Gagen K.; Gorski J.; He S.; Howard A. D.; Jian T.; Jiang M.; Kan Y.; Kelly T. M.; Kosinski J.; Lin L. S.; Liu J.; Marsh D.; Metzger J. M.; Miller R.; Nargund R. P.; Palyha O.; Shearman L.; Shen Z.; Stearns R.; Strack A. M.; Stribling S.; Tang Y. S.; Wang S.-P.; White A.; Yu H.; Reitman M. L. Regulation of Energy Homeostasis by Bombesin Receptor Subtype-3: Selective Receptor Agonists for the Treatment of Obesity. Cell Metabolism 2010, 11, 101–112. [DOI] [PubMed] [Google Scholar]
- Welch C. J.; Gong X.; Schafer W.; Chobanian H. R.; Lin L. S.; Biba M.; Liu P.; Guo Y.; Beard A. Factors Influencing the Interconversion of a New Class of Dibenzodiazepine Sulfonamide Atropisomers. Chirality 2009, 21, 105–109. [DOI] [PubMed] [Google Scholar]
- Barder T. E.; Walker S. D.; Martinelli J. R.; Buchwald S. L. Catalysts for Suzuki-Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure. J. Am. Chem. Soc. 2005, 127, 4685. [DOI] [PubMed] [Google Scholar]
- Chobanian H. R.; Fors B. P.; Lin L. S. A. Facile Microwave-assisted Palladium-catalyzed Cyanation of Aryl Chlorides. Tetrahedron Lett. 47, 3303. [Google Scholar]
- Baker R. K.; Chang L. L.; Chioda M.; Chobanian H. R.; Gao Y.; Guo Y.; Lin L. S.; Liu P.; Nargund R. P.; Rupprecht K. M.; Miao S.. Substituted Diazepine Sulfonamides as Bombesin Receptor Subtype-3 Modulators. PCT Int. Appl. WO 2008073311, 2008.
- Absolute stereochemistry of the benzodiazepine sulfonamide was inferred on the basis of an X-ray structure obtained for compound 1 as previously reported in ref (11).
- See Supporting Information for selected compounds with measured plasma protein binding in human and rat (Table 5).
- See Supporting Information for BRS KO vs WT acute food intake/weight loss study details (Figures 5 and 6).
- AM251 was a cannabinoid 1 receptor antagonist (see: Cannibinoid Receptor (CB1) Antagonist, AM 251, Causes a Sustained Reduction of Daily Food Intake in the Rat. Physiol. Behav. 2004, 82 (5), 863–869) that was used here as a positive control. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.