

Abstract

A structure–activity relationship study of the imidazolyl-β-tetrahydrocarboline series identified MK-4256 as a potent, selective SSTR3 antagonist, which demonstrated superior efficacy in a mouse oGTT model. MK-4256 reduced glucose excursion in a dose-dependent fashion with maximal efficacy achieved at doses as low as 0.03 mg/kg po. As compared with glipizide, MK-4256 showed a minimal hypoglycemia risk in mice.
Keywords: SSTR3, antagonist, type 2 diabetes, β-tetrahydrocarboline
Type 2 diabetes mellitus (T2DM) is a carbohydrate metabolism disorder characterized by high blood glucose levels (hyperglycemia) resulting from the body's deficiency in insulin production and utilization.1 Diabetes has become a worldwide epidemic, affecting 6.4% of the adult population in the developed world. It is predicted that the number affected will grow to 7.7% by 2030.2 Among all of the diabetes cases, more than 90% of the patients have T2DM (noninsulin-dependent diabetes).3 Diabetes causes significant morbidity and mortality. Diabetic retinopathy is the number one cause of preventable blindness in the developed countries, while diabetic kidney disease often leads to end stage renal failure.4,5 Despite the availability of a range of agents for the treatment of T2DM (e.g., sulfonylureas, metformin, PPARγ-selective agonists, and DPP-4 inhibitors), a high proportion of the diabetic patients are undertreated, failing to achieve or maintain glycemic targets. In addition, many current therapies have significant limitations and/or liabilities, particularly in terms of disease modification and durability.6−8 Therefore, effective treatment of T2DM continues to be a huge, unmet medical need.
One strategy for the discovery of safe, effective treatments of T2DM is the development of novel agents that promote glucose-dependent insulin secretion (GDIS) from pancreatic β-cells, thus minimizing the potential risk associated with hypoglycemia. Loss of GDIS from the pancreatic β-cell is a primary defect responsible for the onset and progression of T2DM.9 Oral agents that stimulate insulin secretion, such as sulfonylureas, have proven efficacious in glucose lowering. However, these agents stimulate the β-cell to secrete insulin continuously, regardless of prevailing glucose levels, thereby promoting hypoglycemia and accelerating the loss of islet function.10 Thus, oral agents that induce GDIS have the potential to replace sulfonylureas as a first-line therapy for the treatment of T2DM. To that end, DPP-4 inhibitors represent a major breakthrough such that they selectively enhance insulin secretion in the presence of hyperglycemia by increasing plasma levels of active GLP-1.11,12 DPP-4 inhibitors thus avoid the risk of hypoglycemia commonly seen with sulfonylureas. Novel agents with a GDIS mechanism may offer better efficacy and/or durability, both as monotherapy and in combination.
Somatostatin (also known as growth hormone-inhibition hormone or somatotropin release-inhibiting factor, SRIF) derived its names from its ability to inhibit the release of growth hormone from the anterior pituitary gland.13 Two active forms of somatosatin are produced by alternative cleavage: 14 amino acids (SRIF-14) and 28 amino acids (SRIF-28). Somatostatin also suppresses the production of the pancreatic hormones (e.g., insulin and glucagon), has a role in the central nervous system as a neurotransmitter, is involved in the regulation of gastric secretion, and may regulate cell proliferation. The functions of somatostatin are mediated through five G-protein coupled receptors (SSTR1–SSTR5).
We had found that antagonism of SSTR3 has the potential to be a novel GDIS mechanism for the treatment of T2DM.14 SSTR3 is highly expressed in β-cells of human and rodent islets. Silencing the expression of SSTR3 with siRNA significantly enhanced GDIS in a rat insulinoma cell line, INS-1 cells. The importance of SSTR3 in regulating GDIS and glucose homeostasis was further supported by the fact that a selective SSTR3 antagonist enhanced GDIS in mouse islets and reduced blood glucose levels during a glucose tolerance test (GTT) (Figure 1).15 The glucose lowering was not observed with SSTR3 knockout mice.15 Mechanistically, SSTR3 signals primarily through Gαi-mediated inhibition of cAMP production. Therefore, antagonists of SSTR3 most likely enhance GDIS by increasing intracellular cAMP levels in pancreatic β-cells, a mechanism that is similar to GLP-1 analogues, which is proven to be a glucose-dependent mechanism.16
Figure 1.

Imidazolyl β-carboline SSTR3 antagonists.
Two different structural classes of selective small molecule SSTR3 antagonists have been disclosed, imidazolyl-β-carbolines derived from d-Trp and substituted decahydroisoquinolines.17−18b While both structural classes offer potent, selective SSTR3 antagonists, these compounds have not been reported as potential treatments of T2DM. Starting with the imidazolyl-β-carboline class, compound 1 was found to be a potent and selective SSTR3 antagonist.15 It demonstrated good inhibition (75%) of glucose excursion in the mouse ipGTT (intraperitoneal glucose tolerance test) model at 10 mg/kg oral dose (Table 1). The efficacy was mediated through SSTR3 since the inhibition of glucose excursion was not observed in the SSTR3 knockout mouse. However, 1 has strong binding to hERG as shown in a MK-499 binding assay.19,19b Herein, we describe improved efficacy in this series with a reduced off-target activities (especially hERG) that led to a potential development candidate.
Table 1. Profile of C-1 Monosubstituted Imidazolyl β-Carbolinesa.
| human SSTR3 |
mouse
SSTR3 |
|||||
|---|---|---|---|---|---|---|
| compd no. | binding IC50 (nM) | functional
cAMP antagonism IC50 (nM) (% inh.) |
binding IC50 (nM) | functional
cAMP antagonism IC50 (nM) (% inh.) |
MK-499 binding assay, Ki (nM) | reduction of glucose excursion in mouse ipGTT at 10 mg/kg oral dose (drug level)b,c |
| 1 | 14 | 11 (87%) | 8.9 | 16 (98%) | 369 | 75% (3.8 μM) |
| 2 | 2.8 | 5.7 (119%) | 1.7 | 26 (108%) | 377 | 79% (4.6 μM) |
| 3 | 6.9 | 9.3 (102%) | 4.0 | 4.4 (67%) | 300 | 91% (1.2 μM) |
| 4 | 7.2 | 33 (93%) | 4.8 | 26 (66%) | 2189 | 84% (1.6 μM) |
| 5 | 19 | 19 (102%) | NDd | 18 (103%) | 385 | NDd |
| 6 | 9.2 | 58 (76%) | 8.1 | 29 (92%) | 2206 | 87% (0.2 μM) |
Assay protocols are provided in the Supporting Information. In vitro assay results are the average of at least three duplicates.
Mice were challenged with ip injection of 2 g/kg dextrose.
Drug level refers to the concentration of the compound in plasma 2.5 h postoral dosing at 10 mg/kg.
Not determined.
During SAR optimization of the substitution pattern on the phenyl-imidazolyl ring, the more potent 4-fluorophenyl analogue 2 was synthesized (Table 1). The SSTR3 binding assay measures a compound's competitive displacement of radiolabeled SRIF from cell membranes expressing human or mouse SSTR3 receptor. The SSTR3 antagonist assay is a whole cell functional assay in cells expressing the human or mouse SSTR3 receptor that affords a compound's ability to inhibit the SRIF-induced reduction of cAMP accumulation induced by forskolin. The mouse ipGTT model measures the compound's ability to reduce plasma glucose excursion induced by intraperitoneal injection of dextrose. As compared with 1, compound 2 has improved SSTR3 in vitro potency while maintaining similar MK-499 binding. Both 1 and 2 worked in the mouse ipGTT model, reducing the glucose excursion by 75 and 79%, respectively. Given the enhanced SSTR3 potency, the para-F substitution was incorporated in later analogues.
The 1-(4-tetrahydropyranyl) group was replaced by a variety of heterocycles to afford 3–6. Compounds 3 and 4 are a pair of diastereomers incorporating N-methyl pyrazole at the C1 position of β-tetrahydrocarboline. They have similar SSTR3 binding potency, while 3 is slightly more potent in the SSTR3 functional antagonist assay. Both compounds worked in the mouse ipGTT model with similar plasma drug exposure. However, compound 4 is ∼7-fold less potent in MK-499 binding assay.
Similarly, the diastereomeric pair 5 and 6 with 1,2,4-oxadiazole substitution was prepared. Analogous to the pair of 3 and 4, compounds 5 and 6 have comparable profiles except that diastereomer 6 has less activity in the MK-499 binding assay.
Compounds 4 and 6 were further evaluated in pharmacokinetic (PK) studies (Table 2). In rodents, 6 showed lower clearance and ∼2–3-fold higher exposures after oral dosing than 4. Compound 6 was further tested in higher species and exhibited excellent PK profiles in both dog and rhesus with long half-lives and excellent oral exposures.
Table 2. PK Parameters of 4 and 6.
| mg/kg |
||||||||
|---|---|---|---|---|---|---|---|---|
| compd no. | species | iv dose | oral dose | oral AUC(0–t) (μM h) | iv t1/2 | iv CL (mL/min kg) | Vdss (L/kg) | F (%) |
| 4 | mousea | 4 | 10 | 4.11 | 2.24 | 99.6 | 10.8 | 100 |
| 4 | rata | 1 | 2 | 1.38 | 2.26 | 25.3 | 4.59 | 47 |
| 6 | mousea | 4 | 10 | 7.73 | 2.58 | 39.9 | 7.21 | 76 |
| 6 | rata | 1 | 2 | 4.36 | 8.14 | 9.67 | 5.83 | 58 |
| 6 | dogb | 1 | 2 | 27.6 | 6.69 | 1.78 | 0.87 | 67 |
| 6 | rhesusb | 1 | 2 | 12.4 | 8.08 | 2.79 | 1.75 | 51 |
The compound was dosed as 1.0 mg/mL solution in EtOH:PEG400:water (2:23:75).
The compound was dosed as 4.0 mg/mL solution in EtOH:PEG400:water (10:40:50).
An interesting observation was made with compound 7 where the C1 position has a diphenyl substitution pattern. Compound 7 is more potent than the monosubstituted compounds on SSTR3 in vitro assays (Table 3), but 7 does afford potent binding in MK-499 binding assay.
Table 3. Profiles of Disubstituted Imidazoyl β-Carbolinesa.
| human SSTR3 |
mouse
SSTR3 |
|||||
|---|---|---|---|---|---|---|
| compd no. | binding IC50 (nM) | functional cAMP antagonism IC50 (nM) (% inh.) | binding IC50 (nM) | functional cAMP antagonism IC50 (nM) (% inh.) | MK-499 binding assay, Ki (nM) | reduction of glucose excursion in mouse oGTT at 1 mg/kg oral doseb |
| 7 | 0.76 | 7.3 (91%) | ND | 2.4 (101%) | 44 | NDc |
| 8 | 0.66 | 0.95 (83%) | 0.36 | 0.46 (87%) | 1701 | 109% |
| 9 | 1.1 | 1.1 (107%) | 0.61 | 3.7 (130%) | 1638 | 63% |
| 10 | 2.3 | 3.6 (77%) | 1.2 | 4.8 (92%) | 462 | 81% |
| 11 | 0.94 | 0.78 (75%) | 0.50 | 0.14 (68%) | 543 | 80% |
| 12 | 0.74 | 0.70 (103%) | 0.46 | 0.48 (113%) | 2616 | 68% |
| 13 | 0.97 | 1.2 (69%) | 0.59 | 1.3 (133%) | 1403 | 71% |
Assay protocols are provided in the Supporting Information. In vitro assay results are the average of at least three duplicates.
Mice were challenged with oral dose of 5 g/kg dextrose.
Not determined.
The enhanced potency of 7 prompted further investigation of compounds with disubstitution at the C-1 position (Table 3). The N-methyl pyrazole and 1,2,4-oxadiazole of compounds 4 and 6 were combined to afford potent isomers 8 and 9. Both compounds maintain excellent in vitro potency and reduced activity in the MK-499 binding assay. Screening in the original mouse ipGTT model was replaced with an oral glucose tolerance test (oGTT) due to the ease of the experimental protocols and analogy to oGTT protocols carried out in human clinical trials. Compound 9 reduced glucose excursion by 63% at 1 mg/kg po, while compound 8 achieved complete ablation of glucose excursion (109%) at the same dose.
The enantiomers 10 and 11 were also synthesized, starting from d-Trp. Interestingly, compounds 10 and 11 have similar SSTR3 potency as compounds 8 and 9. However, they suffer from increased binding to hERG. Compounds 12 and 13, which have two identical heterocycles moieties at the C1 position, were also synthesized. Although both compounds maintain high SSTR3 in vitro potency, and they were less efficacious in the mouse oGTT model than compound 8 following oral dosing at 1 mg/kg.
Synthesis of all compounds was based on the Pictet–Spengler cyclization of the phenyl imidazolyl tryptamine with the corresponding aldehydes and ketones.17−17c,20 The reaction with aldehydes was a mild process, occurring at room temperature, while the reaction with ketones required more vigorous conditions (Scheme 1).
Scheme 1. Synthesis of Imidazoyl Carboline,

Experimental details are included in the Supporting Information.
Reaction conditions: CH2Cl2, TFA, RT for aldehyde; pyridine, 70–100 °C for ketone.
With the excellent efficacy of 8, the compound was titrated in the mouse oGTT assay (Figure 2). In this graph, the black bar represents the glucose excursion when the mice were challenged orally with 5 g/kg dextrose. It was clear that compound 8 reduces the glucose excursion from 0.003 to 10 mg/kg in a dose-dependent manner. The plasma Cmax of 8 was determined from parallel mouse PK studies. At 0.01, 0.1, and 1 mg/kg oral dose, compound 8 achieved Cmax of 7, 88, and 493 nM, respectivley.
Figure 2.
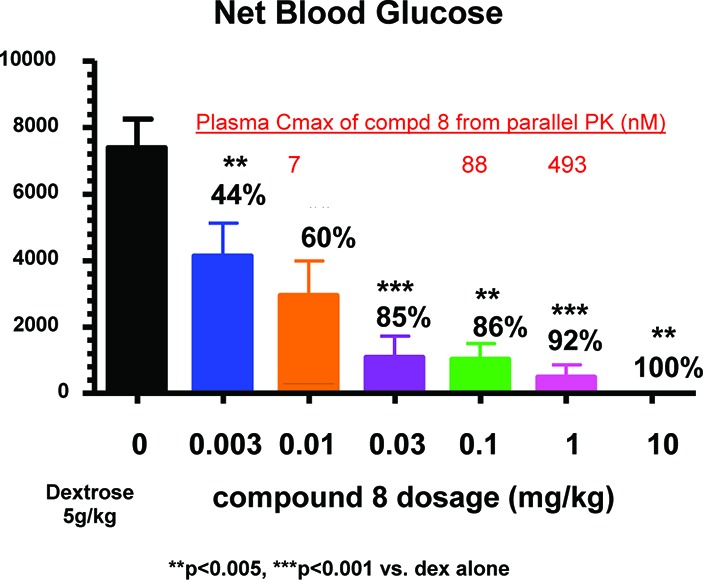
Compound 8 dose dependently reduces glucose excursion in the mouse oGTT model.
To demonstrate that the observed glucose lowering by compound 8 was SSTR3-dependent, the effect of a maximally efficacious dosage of 8 on blood glucose excursion during an oGTT was investigated in SSTR3 KO mice (Figure 3). Administration of 8 (1 mg/kg) and compound A (1 mg/kg; des-F-sitagliptin, a DPP-4 inhibitor included as a positive control) to age-matched C57BL/6N male WT mice significantly inhibited blood glucose excursion by 112 and 91%, respectively. When equivalent dosages were administered to SSTR3–/– mice, 8 reduced glucose excursion by 28% (not significant), while compound A retained its efficacy as expected (reduced glucose by 109%).
Figure 3.
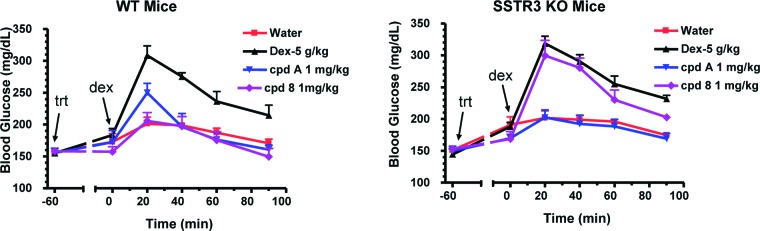
Effects of 8 and DPP-4 inhibitor compound A (des-F sitagliptin) on oGTT glucose levels in SSTR3 KO and WT mice.
Compound 8 has excellent selectivity against other SSTR subtypes based on in vitro assays (Table 5). In human receptor binding assays, 8 has IC50 values >2 μM for SSTR1 and SSTR2. Although the binding IC50 values on SSTR4 and SSTR5 are below 1 μM, there is still >500-fold selectivity. Compound 8 was tested in functional antagonist assays against SSTR4 and SSTR5. The IC50 values are greater than 5 μM (at least 5000-fold selectivity). Compound 8 was also tested in functional agonist assays against SSTR2 to 5. The data indicate that compound 8 is not an agonist against these receptor subtypes.
Table 5. SSTR Subtype Selectivity of Compound 8a.
| human functional cAMP |
|||
|---|---|---|---|
| receptor | human binding IC50 (nM) | antagonism IC50 (nM) (% inh. at max. dose) | agonism IC50 (nM) (% act. at max. dose) |
| SSTR1 | 2362 | NDb | NDb |
| SSTR2 | 4025 | NDb | –9% at 5 μMc |
| SSTR3 | 0.66 | 0.95 (83% at 2 μM) | 6% at 20 μMc |
| SSTR4 | 384 | 42% at 20 μMc | 27% at 5 μMc |
| SSTR5 | 533 | 34% at 5 μMc | 6% at 20 μMc |
Assay protocols are provided in the Supporting Information. In vitro assay results are the average of at least three duplicates.
Not determined.
IC50 was not calculated.
Compound 8 was further characterized in PK studies in preclinical species (Table 4). It has excellent oral bioavailability across species with long half-lives and high drug exposures in higher species (dog and monkey).
Table 4. Pharmacokinetic Parameters of 8.
| mg/kg |
|||||||
|---|---|---|---|---|---|---|---|
| species | iv dose | oral dose | oral AUC(0–t) (μM h) | iv t1/2 | iv CL (mL/min kg) | Vdss (L/kg) | F (%) |
| mousea | 4 | 10 | 19.3 | 1.52 | 10.1 | 1.66 | 58 |
| ratb | 1 | 2 | 0.81 | 1.73 | 34.0 | 5.10 | 42 |
| dogc | 1 | 2 | 15.6 | 7.00 | 2.34 | 1.52 | 53 |
| rhesusc | 1 | 2 | 14.4 | 7.21 | 3.09 | 1.74 | 65 |
The compound was dosed as 1.0 mg/mL solution in EtOH:PEG400:water (2:23:75).
The compound was dosed as 1.0 mg/mL solution in EtOH:PEG400:water (20:30:50).
The compound was dosed as 4.0 mg/mL solution in EtOH:PEG400:water (10:40:50).
A single crystal X-ray of compound 8 was obtained to allow unambiguous structure determination (Figure 4). The structure confirmed that compound 8 has the R,R configuration at the two stereogenic centers, C-1 and C-3. It is interesting to note that the β-tetrahydrocarboline and phenyl-imidazolyl groups of 8 adopt a near-planar conformation with the two hetereocycles pointing up or down.
Figure 4.
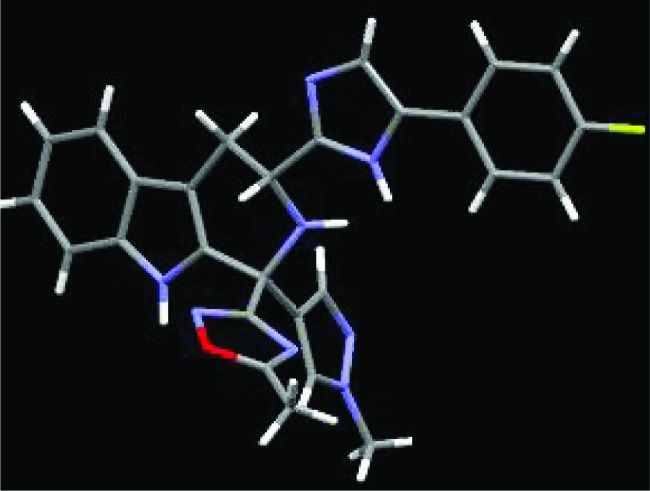
X-ray structure of 8 (toluene solvent deleted for clarity).
Compound 8 was also evaluated for its potential to induce hypoglycemia in lean C57BL/6N mice (Figure 5). After a 4 h fast, basal blood glucose concentrations averaged 161 mg/dL. Compound 8 was administered at 0.01, 0.1, and 1 mg/kg by gavage. Blood glucose levels did not decrease significantly at all three doses over the subsequent 5 h. In contrast, glipizide, a marketed sulfonylurea K+-ATP blocker, dosed at 10 mpk, caused significant decreases of blood glucose levels, starting at 60 min after dosing of glipizide.
Figure 5.
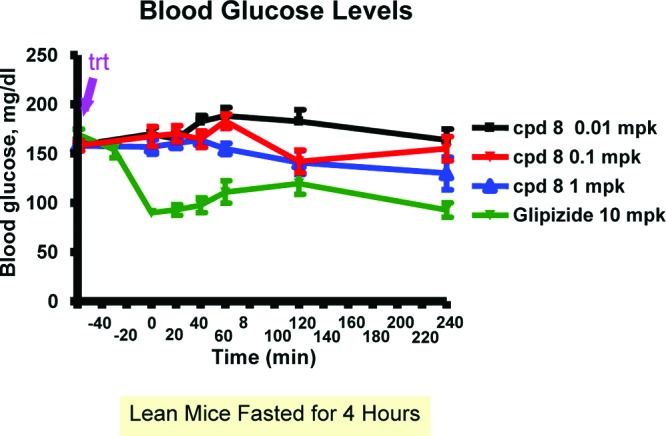
Compound 8 has minimal hypoglycemia risk.
In conclusion, we discovered compound 8 (MK-4256) as a potent antagonist of SSTR3. MK-4256 demonstrated exceptional SSTR3-mediated glucose-lowering efficacy in the mouse oGTT model with minimal hypoglycemia risk. When combined with a DPP-4 inhibitor, there is significant synergy in glucose-lowering efficacy.21 MK-4256 was selected for further development as a treatment for T2DM.22 Additional data will be reported in due course.
Acknowledgments
We thank Dr. Charles W. Ross III at Merck Research Laboratories for measuring the high resolution mass of 8 (MK-4256).
Supporting Information Available
Syntheses and characterization data for compounds 2–13 and assay protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Del Prato S.; Marchetti P.; Bonadonna R. C. Phasic Insulin Release and Metabolic Regulation in Type 2 Diabetes. Diabetes 2002, 51Suppl. 1S109–S116. [DOI] [PubMed] [Google Scholar]
- Shaw J. E.; Sicree R. A.; Zimmet P. Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [DOI] [PubMed] [Google Scholar]
- Gonzalez E. L.; Johansson S.; Wallander M. A.; Rodriguez L. A. Trends in the prevalence and incidence of diabetes in the UK: 1996–2005. J. Epidemiol. Community Health 2009, 63, 332–336. [DOI] [PubMed] [Google Scholar]
- Scanlon P. H. The English national screening programme for sight-threatening diabetic retinopathy. J. Med. Screen. 2008, 1511–4. [DOI] [PubMed] [Google Scholar]
- Palmer A. J.; Valentine W. J.; Chen R.; Mehin N.; Gabriel S.; Bregman B.; Rodby R. A. A health economic analysis of screening and optimal treatment of nephropathy in patients with type 2 diabetes and hypertension in the USA. Nephrol., Dial., Transplant. 2008, 23, 1216–1223. [DOI] [PubMed] [Google Scholar]
- Petrie J. R.; Adler A.; Vella S. What to add in with metformin in type 2 diabetes?. Q. J. Med. 2011, 104, 185–192. [DOI] [PubMed] [Google Scholar]
- Kokil G. R.; Rewatkar P. V.; Verma A.; Thareja S.; Naik S. R. Pharmacology and Chemistry of Diabetes mellitus and Antidiabetic Drugs: A Critical Review. Curr. Med. Chem. 2010, 17, 4405–4423. [DOI] [PubMed] [Google Scholar]
- Krentz A. J.; Patel M. B.; Bailey C. J. New drugs for type 2 diabetes mellitus: What is their place in therapy?. Drug 2008, 68152131–2162. [DOI] [PubMed] [Google Scholar]
- Butler A. E.; Janson J.; Bonner-Weir S.; Ritzel R.; Rizza R. A.; Butler P. C. β-Cell Deficit and Increased β-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes 2003, 521102–110. [DOI] [PubMed] [Google Scholar]
- Bodmer M; Meier C.; Krähenbühl S.; Jick S. S.; Meier C. R. Metformin, Sulfonylureas, or Other Antidiabetes Drugs and the Risk of Lactic Acidosis or Hypoglycemia, A nested case-control analysis. Diabetes Care 2008, 31112086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A. E. Dipeptidyl Peptidase IV Inhibitors for the Treatment of Diabetes. J. Med. Chem. 2004, 47, 4135–4141. [DOI] [PubMed] [Google Scholar]
- Deacon C. F. Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes: a comparative review. Diabetes, Obes. Metab. 2011, 13, 7–18. [DOI] [PubMed] [Google Scholar]
- Olias G.; Viollet C.; Kusserow H.; Epelbaum J.; Meyerhof W. Regulation and function of somatostatin receptors. J. Neurochem. 2004, 89, 1057–1091. [DOI] [PubMed] [Google Scholar]
- Li J.; Zhou Y.; et al. Manuscript in preparation.
- Pasternak A.; Feng Z.; deJesus R.; Ye Z.; He S.; Dobbelaar P. H.; Bradley S. A; Chicchi G. G.; Tsao K.; Trusca D.; Eiermann G. J.; Feng Y.; Wu M.; Shao Q.; Zhang B.; Nargund R. P.; Mills S. G.; Howard A. D.; Yang L.; Zhou Y. Stimulation of Glucose-Dependent Insulin Secretion by a Potent, Selective sst3 Antagonist. ACS Med. Chem. Lett. 2012, 3, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrén B. GLP-1 for type 2 diabetes. Exp. Cell Res. 2011, 317, 1239–1245. [DOI] [PubMed] [Google Scholar]
- Poitout L.; Roubert P.; Contour-Galcera M.; Moinet C.; Lannoy J.; Pommier J.; Plas P.; Bigg D.; Thurieau C. Identification of Potent Non-Peptide Somatostatin Antagonists with sst3 Selectivity. J. Med. Chem. 2001, 44, 2990. [DOI] [PubMed] [Google Scholar]
- Thurieau C.; Poitout L.; Galcera M.; Moinet C; Gordon T. D.; Morgan B. A.; Bigg D.; Pommier J.. β-Carboline Compounds. WO 1999/64420, December 16, 1999.
- Troxler T.; Hurth K.; Hoyer D.. Beta-Carboline Derivatives and Its Pharmaceutical Use Against Depression and Anxiety. WO 2002/081471, April 2, 2002.
- Troxler T.; Hurth K.; Schuh K.; Schoeffter P.; Langenegger D.; Enz A.; Hoyer D. Decahydroisoquinoline derivatives as novel non-peptidic, potent and subtype-selective somatostatin sst3 receptor antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 1728–1734. [DOI] [PubMed] [Google Scholar]
- Baenziger M.; Cercus J.; Hirt H.; Laumen K.; Malan C.; Spindler F.; Struber F.; Troxler T. The development of a practical synthesis of the potent and selective somatostatin sst3 receptor antagonist [4-(3,4-difluoro-phenyl)-piperazine-1-yl]-{(4S,4aS,8aR)-2[(S)-3-(6-methoxy-pyridin-3-yl)-2-methyl-propyl]-decahydroisoquinoline-4-yl}-methanone (NVP-ACQ090). Tetrahedron: Asymmetry 2003, 14, 3469–3477. [Google Scholar]
- Radiolabeled MK-499 has been used in a hERG (potassium ion channel Kv11.1) binding assay to provide an initial screen of a compound's potential to interact with hERG. Blocking hERG can result in QTc prolongation, a serious adverse effect.; Raab C. E.; Butcher J. W.; Connolly T. M.; Karczewski J.; Yu N. X.; Staskiewicz S. J.; Liverton N.; Dean D. C.; Melillo D. G. Synthesis of the first sulfur-35-labeled hERG radioligand. Bioorg. Med. Chem. Lett. 2006, 16, 1692–1695. [DOI] [PubMed] [Google Scholar]
- Wang J.; Della Penna K.; Wang H.; Karczewski J.; Connolly T. M.; Koblan K. S.; Bennett P. B.; Salata J. J. Functional and pharmacological properties of canine ERG potassium channels. Am. J. Phys 2003, 284, H256–H267. [DOI] [PubMed] [Google Scholar]
- He S.; Lai Z.; Yang D. X.; Hong Q.; Reibarkh M.; Nargund R. P.; Hagmann W. K. Synthesis of β-carboline from aldehydes and ketones via the α-siloxy-α,β-unsaturated esters. Tetrahedron Lett. 2010, 51, 4361–4364and references therein. [Google Scholar]
- Li C.; et al. Manuscript in preparation.
- Process chemistry for the first GMP delivery of MK-4256 has been disclosed recently:Ruck R. T.; Huffman M. A.; Stewart G.; Cleator E.; Kandur W. A.; Kim M. M.; Zhao D.. Process chemistry for the first GMP delivery of SSTR3 antagonist MK-4256. Abstracts of Papers, Presentation (ORGN-461), 243rd ACS National Meeting, San Diego, CA, March 25–29, 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.