

Abstract

Tri- and tetracyclic nitrogen-bridgehead compounds were designed and synthesized to yield micromolar cholinesterase (ChE) inhibitors. Structure–activity relationships identified potent compounds with butyrylcholinesterase selectivity. These compounds were selected as starting points for the design and synthesis of carbamate-based (pseudo)irreversible inhibitors. Compounds with superior inhibitory activity and selectivity were obtained and kinetically characterized also with regard to the velocity of enzyme carbamoylation. Structural elements were identified and introduced that additionally showed neuroprotective properties on a hippocampal neuronal cell line (HT-22) after glutamate-induced intracellular reactive oxygen species generation. We have identified potent and selective pseudoirreversible butyrylcholinesterase inhibitors that release reversible inhibitors with neuroprotective properties after carbamate transfer to the active site of cholinesterases.
Keywords: Alzheimer's disease, butyrylcholinesterase, carbamates, neuroprotection
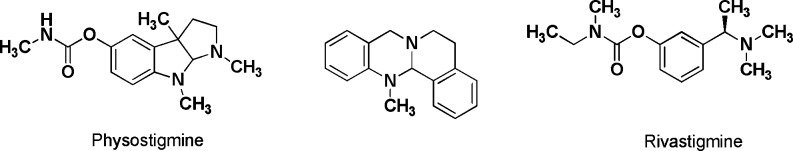
Alzheimer's disease (AD) is one of the most often occurring and most devastating neurodegenerative diseases. Huge efforts have been made to investigate the pathophysiology of AD, but to make a long story short, the disease is still incurable.1 The number of approved drugs is extremely limited to one NMDA antagonist (memantine) and three acetylcholinesterase (AChE) inhibitors (rivastigmine, donepezil, and galantamine). Despite their purely symptomatic mode of action to improve cognition and memory, their clinical effectiveness was proven in a number of studies.2 While galantamine and donepezil are reversible cholinesterase (ChE) inhibitors, rivastigmine (Exelon) represents an irreversible inhibitor transferring a carbamate moiety to the serine unit in the active center of AChE and butyrylcholinesterase (BChE), respectively, the second cholinesterase in the human body.3,3b The carbamates' mode of action is termed pseudoirreversible since the carbamate moiety slowly hydrolyses from the enzyme, yielding the active enzyme, again in contrast to irreversibly inhibiting organophosphates. Rivastigmine, a compound derived from the alkaloid physostigmine, is of special interest because in clinical and pharmacological investigations, it was shown to be of high effectiveness, which some authors attributed to its ability to inhibit also BChE with high efficiency (Chart 1).3b,4 There has been postulated a putative beneficial role in the therapy of AD through BChE inhibition; for example, it was found that the amount of AChE decreases in later stages of AD (when AChE inhibitors become clinically ineffective), whereas the amount of BChE stays the same or is even increased.5,5b Also, it was shown that with a decreasing amount of AChE, BChE seems to be able to compensate for AChE loss, resulting in improved cognition.5b BChE's role in the human body is not yet fully understood; it is responsible for detoxification of xenobiotics, and it influences lipoprotein metabolism and may also play a role in neuronal differentiation.3−5b
Chart 1. Structures of the ChE Inhibiting Alkaloid Physostigmine, the Tetracyclic Lead Structure (5a, cf. Scheme 1), and the AD Drug Rivastigmine.
For the benefit of developing more effective AD drugs and to investigate BChE's physiological functions, there have been efforts in developing selective BChE inhibitors. Gilmer's group has described a very interesting class of pseudoirreversible carbamate inhibitors based on an isosorbide template with remarkable potency and selectivity.6 Our group has used two alkaloids (deoxyvasicine and dehydroevodiamine) that had been described as moderately active and unselective ChE inhibitors as lead structures to identify a series of N-bridgehead tri- and tetracyclic compounds as reversible and competitive inhibitors with improved inhibitory activities and BChE selectivity (Chart 1).7,7b The greatest BChE selectivities were achieved with quinazolinimine structures.7b By applying the bivalent compound approach and the design of hybrid molecules, their pharmacological activities were further improved and enlarged.8−9b
Because even the best ChE inhibitors stay symptomatically acting compounds, there has been considerable interest in applying antioxidant compounds as disease-modifying drugs in AD drug development. Oxidative stress and the formation of reactive oxygen species (ROS) are key processes in AD and result in neuronal damage and subsequent cell death.10,10b It was therefore tried to combine AChE inhibitors with compounds possessing antioxidant properties, for example, by forming covalently connected hybrids or physiologically more labile codrugs.8,8b,11,11b Albeit highly promising compounds, hybrids also suffer from some difficulties, such as their inherent high molecular mass and therefore reduced bioavailability.8a
The vast majority of recent work on cholinesterase inhibitors, mainly hybrids, has been done on the basis of a surprisingly small number of chemical templates, especially tacrine.8b There is a constant search for novel structural templates for ChE inhibitors, especially from natural products.12−12c Our rationale in this present work was to use a previously described tetracyclic lead structure as a template for the development of novel pseudoirreversible inhibitors in which the heterocyclic structure serves as a kind of carrier into the active center of ChEs. Such compounds would release reversible inhibitors after transfer of the carbamate moiety to the enzymes, ideally enabling a more pronounced efficiency of inhibition. Structure–activity relationships (SARs) were investigated for the heterocyclic template, in particular to find out whether the introduction of a phenolic hydroxyl group to enable connection with a carbamate unit is tolerated. Additionally, SARs of the resulting carbamates with the aim to achieve higher selectivity and inhibitory activity toward BChE were investigated. A second rationale of this work was to incorporate antioxidant properties into our target compounds, but not by connecting them covalently to antioxidant moieties as it is the case for hybrid molecules, but to use the heterocyclic phenolic structure of the reversible inhibitor itself as an antioxidant. Weinstock and Groner have used such an approach by combining carbamate structures with MAO-B inhibitors.13 Phenolic structures are key factors for antioxidant and neuroprotective properties in a number of natural products, such as quercetin, ferulic acid, and silibinin.11,11b
We assumed that the compounds released after carbamate transfer represent antioxidants (since, for example, cyclic quinonimines could be formed after oxidation) and could therefore enlarge the biological spectrum of the compounds without increasing their molecular mass. For assaying antioxidant properties, we used two different assays: the first one, the oxygen radical absorbance capacity (ORAC) assay, determines the ability of compounds to protect fluorescein from destruction by peroxyl radicals and, therefore, directly assesses their antioxidant physicochemical properties.9b,11b The second assay is cell-based and uses murine HT-22 hippocampal cells that lack ionotropic glutamate receptors to study intracellular oxidative stress-induced neurotoxicity.11,14,14b In this neuronal oxidative stress-induced toxicity (oxytosis), cell death is induced by extracellular glutamate challenge in a nonreceptor-mediated “oxidative” pathway. Glutamate toxicity is mediated by the cystine/glutamate antiporter system, which—after exposure to high extracellular glutamate concentrations—results in inhibition of cystine uptake, leading to intracellular cysteine and therefore glutathione depletion, which induce ROS accumulation and cell injuries.14−15 Administration of antioxidants such flavonoids can effectively prevent oxidative neuronal death in this cell line.16,16b
For the investigation of SARs concerning the tri- and tetracyclic N-bridgehead compounds, the six-membered ring system containing compound 3,4-dihydroisoquinoline and the seven-membered one 4,5-dihydro-3H-benzo[c]azepine were reacted with isotoic acid anhydride, a reactive form of anthranilic acid formed after reaction with triphosgene (Scheme 1). Preparation of isotoic acid anhydrides also provides an easy method for selective monomethylation of anthranilic acid's amino group. Seven- and six-membered rings were synthesized to investigate their influence on BChE selectivity (previous work on related quinazolinimines showed higher selectivity with increasing ring size).7b,9,9b
Scheme 1. Synthesis of Tetra- and Tricyclic N-Bridgehead Compounds.

Reagents: (i) (Cl3CO)2CO, dry THF, 40–50 °C, 3 h. (ii) CH3I, N,N-diisopropylethylamine, N,N-dimethylacetamide, 40 °C, 24 h. (iii) Toluene/reflux, 24 h. (iv) LiAlH4, THF, 70 °C, 3 h. (v) Ethyl(methyl)carbamic chloride, NaH, THF, rt or isocyanate, Et3N, CH2Cl2, rt. (vi) Pyrrolidin-2-one, MW 100–200 W, 130 °C, 1 h. (vii) Benzylbromide, K2CO3, acetone, 70 °C, 24 h. (viii) CH3I, dioxane, 90 °C, 24 h. (ix) LiAlH4, THF, 70 °C, 3 h. (x) H2, Pd/C, ethanol, rt, 24 h. (xi) Isocyanate, CH2Cl2, Et3N, rt, 1–3 h.
Reduction of the amide group of compounds 3a–c and 4a,c by lithium aluminum hydride afforded the desired tetracyclic quinazoline compounds 5a–c and 6a,c, respectively. Compound 6b was synthesized by demethylation of 6c using boron tribromide. Compound 5b was treated with ethyl(methyl)carbamic chloride in the presence of sodium hydride to afford 8a in 60% yield. The reaction of n-heptylisocyanate with 6b in the presence of sodium hydride gave 7 in 75% yield. Carbamoylation of 5b with the appropriate isocyanate in dichloromethane in the presence of triethylamine gave the carbamates 8b–e in good yields (72–88%) (Scheme 1). Unsubstituted tetracyclic compounds 5a and 6a were prepared, and compounds bearing a methoxy (5c and 6c) and a hydroxyl group (5b and 6b) to test whether substitution in this position is tolerated. Determination of IC50 values at AChE and BChE confirmed that the introduction of −OCH3 or −OH groups has no influence on the inhibitory activities and, therefore, proved that this position of the heterocycle is ideal for introduction of the carbamoyl unit (Table 1). Interestingly, for the seven-membered ring-containing compounds, the inhibition data were considerably modified: As expected from the results with tricyclic quinazolinimines, activity at BChE was slightly higher, whereas inhibitory activities at AChE were slightly lower, resulting in increased selectivity toward BChE (>8).7b Again, introduction of a methoxy or hydroxyl group did not result in a significant alteration of binding behavior. It has to be stated though that all compounds are only 1–2 digit micromolar inhibitors of BChE, and selectivities were never higher than 10.
Table 1. AChE and BChE Inhibition Results, Resulting ChE Selectivity, and Antioxidant Capacities Expressed as Trolox Equivalents (Concentration Range, 0.5–7.5 μM).

AChE from electric eel and BChE from equine serum; data are the means of at least three independent determinations.
Selectivity ratio [IC50(AChE)/IC50(BChE)].
Inhibition at 100 μM, 26% inhibition.
21% inhibition.
33% inhibition.
Concentration range, 1.0–5.0 μM.
8% inhibition.
18% inhibition.
1% inhibition.
24% inhibition.
We also synthesized analogous tricyclic quinazoline compounds to investigate their binding profiles. Their synthesis was accomplished as illustrated in Scheme 1. The fusion of isatoic anhydrides 1a,b with pyrrolidin-2-one using a microwave reactor at 100–200 W power and 100 °C temperature afforded 9a,b in good yields (79–80%). Benzylation of 9b by benzylbromide in the presence of K2CO3 afforded 9c. Quaternization reaction of 9a,c with methyl iodide gave 10a,c. Reduction of 10a,c by lithium aluminum hydride afforded the desired tricyclic quinazoline compounds 11a,c in good yields (74–78%). Debenzylation reaction of 11c gave 12. Finally, carbamoylation of 12 to yield the different carbamates 13a–d was accomplished as reported for the preparation of 8b–e in good yields (68–83%) (Scheme 1).
For investigation into the SARs of this class of compounds, several structurally diverse carbamate groups were introduced at the phenolic hydroxyl group. Several aliphatic and aromatic moieties were used in previous work on carbamates: the methyl ethyl carbamate occurs in rivastigmine (Chart 1) and, therefore, served as our first structure. Previous medicinal research has been done using a physostigmine or rivastigmine-based heterocyclic core:17,17b The compounds synthesized inhibited both ChEs in the nanomolar range. A heptyl group was identified that led to high acitivity, in the case of conformationally restricted rivastigmine analogues with 16-fold selectivity [heptylphysostigmine (eptastigmine) shows 4-fold selectivity].17b The synthesis of phenylcarbamates has been described, again connected to a physostigmine-related heterocyclic core.17b In this case, modulation of selectivity could be achieved by fairly minor structural modifications, for example, a methyl group in the o-position led to AChE selectivity, whereas an isobutyl substituent in the p-position led to BChE selectivity.17a We therefore also applied these carbamate structures to our heterocyclic templates to investigate SARs.
Tri- as well as tetracyclic target compounds bear a chiral C atom attached to two N atoms, but at this stage, no enantioseparation was performed. The stability of carbamoylated compounds under assay conditions was investigated by incubation of 8a and 8b in buffer and control by LC-ESI-MS, and no decomposition was observed.
The tetracyclic carbamate compound 8a bearing methyl and ethyl substituents at the carbamate group (like rivstigmine) shows significant improvement in binding activities over noncarbamate compounds, moderately pronounced at AChE (6-fold more potent as compared to the unsubstituted compound 5a) and very pronounced at BChE (100-fold more potent as compared to the unsubstituted compound 5a), leading to submicromolar activities at BChE and 54-fold selectivity over AChE (Table 1). The introduction of a heptyl substituent as in heptylphysostigmine for compounds 7 and 8b lead to a further 10-fold increase in activity at BChE and a very remarkable decrease in activity at AChE. For compound 8b, no activity at all could be observed even at 100 μM. So, two-digit nanomolar inhibitors of BChE were obtained lacking any activity at AChE. The tricyclic heptyl carbamate 13a only showed moderate activity and selectivity with IC50 (AChE) = 32.3 μM and IC50 (BChE) = 2.0 μM.
We also studied the effect of phenylcarbamates on ChE inhibition activity and selectivity. Tetracyclic 3-methoxyphenyl-, 2-methylphenyl-, and 4-isopropylphenylcarbamates 8c, 8d, and 8e, respectively, showed submicromolar activities at BChE with 900–1500-fold selectivity over AChE (Table 1). Tricyclic 3-methoxyphenyl-, 2-methylphenyl-, and 4-isopropylphenyl carbamates 13b, 13c, and 13d, respectively, only showed moderate activity and selectivity with IC50 (AChE) ranging from 40 to lower than 200 μM and IC50 (BChE) ranging from 3 to 8 μM (Table 1).
Kinetic studies were performed to get more detailed information about the time course of inhibition and therefore more precise data about the equilibrium constant of inhibition (for experimental details, cf. Supporting Information). The equilibrium constant of the inhibitor–ChE complexes (KC) and the carbamoylation rate constants (k3) of two representative target compounds 8a and 8b and physostigmine as a positive control were determined (Figure 1). The KC value calculated for physostigmine as positive control is in agreement with the value reported in the literature.17b For compound 8a at AChE, the actual KC value is even smaller than determined as the IC50 values with KC = 1.1 μM and at BChE with KC = 274 nM, resulting in 4-fold selectivity. The rate constants of 0.31 min–1 (AChE) and 0.18 min–1 (BChE) show rapid carbamoylation, albeit slower than for physostigmine (Figure 1). For compound 8b, KC (BChE) is even lower as for determination of the IC50 value with KC = 12.7 nM, and no carbamoylation could be observed at AChE for the concentrations tested. The velocity of carbamoylation was fairly rapid with k3 = 0.15 min–1. Compound 8b shows a time-dependent pattern of inhibition characterized by an increase until a steady state after 60 min (Figure 1).
Figure 1.
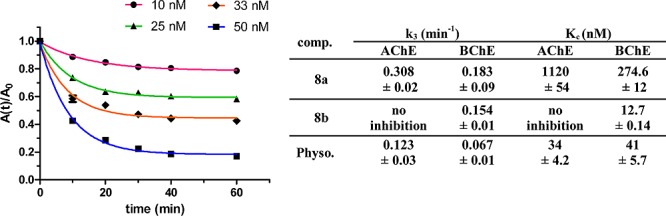
(A) Time-dependent pattern of inhibition of BChE by compound 8b (10–50 nM). (B) Stability constants of the inhibitor–ChE complex (KC) and rate constants of carbamoyl–ChE formation (k3) of 8a,b and physostigmine, respectively.
The tetracyclic heptyl carbamate 8b represents a highly potent pseudoirreversible BChE inhibitor with KC = 12.7 nM without any activity at AChE and fast onset of action.
For evaluation of antioxidant potencies, in a first assay, the target compounds' radical scavenging capacities were determined by means of their ability to reduce the amount of peroxyl radicals (ORAC assay). Results are expressed in “trolox” (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, a water-soluble vitamin E derivative) equivalents.9b As can be seen from Table 1, the phenolic compounds (5b, 6b, and 12) are by far the most potent compounds with antioxidant capacities ranging from 2.5 to 3.4 trolox equivalents, meaning that they are far more potent than the very active positive control trolox. The methoxy compounds 5c and 6c show lower potencies with 0.5 and 1.2 trolox equivalents, respectively. The unsubstuituted compound 5a is among the least potent compounds. Finally, the carbamates tested show only very moderate antioxidant properties (0.65 trolox equiv for the ethyl methyl carbamate 8a, 0.30 trolox equiv for the tetracyclic heptyl carbamate 8b, and 1.03 equiv for the tricyclic heptyl carbamate 13a). None of these compounds lacks radical scavenging properties completely, though, and for structurally related compounds, there can be significant differences in their capacities.9b As a general rule, it can be seen that the hydroxyl compounds released after carbamate transfer to the enzymes' serine unit represent highly potent antioxidants with slightly higher capacities for the tricyclic compound (Table 1).
In a second assay assessing the target compounds' antioxidant properties, their ability to prevent oxycytotic cell death of murine hippocampal HT-22 cells after glutamate exposure and subsequent intracellular ROS formation was evaluated. The six- and seven-membered ring tetracyclic hydroxyl compound 5b and 6b, as well as the tricyclic hydroxyl-compound 12 formed after carbamate transfer to the enzyme, showed very pronounced neuroprotective properties (tested at 10 μM) to the same extent as the potent positive control quercetin (tested at 25 μM) and much stronger than the flavolignan silibinin.11a The six-membered ring heterocyclic methoxy compound 5c showed activity in the same range, although performing these experiments at various concentrations revealed that the hydroxy compound exhibits neuroprotection already at 5 μM, when the methoxy compound is not active (cf. Supporting Information for data at 5 μM). The unsubstituted compound 5a showed no activity. Surprisingly, all tested carbamates 7, 8b, and 13a showed potent antioxidant capacities to the same extent as quercetin does, and only the tricyclic compound 13a showed slightly lower activities at a concentration of 5 μM. The methyl ethyl-substituted carbamate 8a did not show neuroprotective activity at 10 μM (Figure 2).
Figure 2.

Evaluation of neuroprotection of unsubstituted, phenolic, methoxy-substituted, and selected carbamyolated compounds at 10 μM against glutamate-induced oxidative stress on HT-22 cells (cf. the Supporting Information for details).
The phenolic hydroxyl group, which is also formed after carbamate transfer to the enzyme, led to potent ROS scavenging molecules exhibiting up to 3.5 trolox equiv in a physicochemical ORAC assay. Interestingly, evaluation of the antioxidant properties of the target compounds on glutamate-challenged neuronal HT-22 cells (and therefore increased intracellular ROS production) showed very potent neuroprotective properties not only for the hydroxyl compounds but also for the carbamates in the same or higher range than for the positive control quercetin. These results are surprising since we expected high neuroprotection only for the hydroxyl compounds, but obviously, in the cell-based assay also, the carbamate compounds are very potent neuroprotectants in their own regard.
Into a novel heterocyclic template consisting of basic tri- and tetracyclic N-bridgehead compounds that inhibit ChEs, a phenolic hydroxyl group was introduced, and SARs showed that the −OH group as well as respective phenol ethers are tolerated without loss of activity. Introduction of carbamate units not only increased inhibitory activities into the submicromolar range, but SARs on the carbamate moiety led to nanomolar inhibitors with ≫15000-fold selectivity toward BChE over AChE. Kinetic studies on selected carbamates proved pseudoirreversible inhibition and—depending on the concentration of inhibitors—a comparatively fast onset of action. Kinetic studies revealed an even higher stability constant (KC) for compounds 8a and 8b as reflected by the IC50 value alone.
We are currently performing computational and docking studies to explain the very pronounced changes in enzyme inhibitory activity and selectivity by only minor changes in the chemical structure, and results will be presented at a later stage.
Acknowledgments
A Ph.D. scholarship of the German Academic Exchange Service (DAAD) for F.H.D. is gratefully acknowledged. Appreciation is expressed to Gabriele Brunner for technical assistance, to J. Kiermeier for MS measurements, and to Professor S. Elz for scientific mentoring.
Glossary
Abbreviations
- AD
Alzheimer's disease
- AChE
acetylcholinesterase
- BChE
butyrylcholinesterase
- ROS
reactive oxygen species
- ORAC
oxygen radical absorbance capacity
Supporting Information Available
Elemental analysis and spectral data, detailed synthetic and pharmacological procedures, and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Ballard C.; Gauthier S.; Corbett A.; Brayne C.; Aarsland D.; Jones E. Alzheimer's disease. Lancet 2011, 377, 1019–1031. [DOI] [PubMed] [Google Scholar]
- Takeda A.; Loveman E.; Clegg A.; Kirby J.; Picot J.; Payne E. C. A systematic review of the clinical effectiveness of donepezil, rivastigmine and galantamine on cognition, quality of life and adverse events in Alzheimer's disease. Int. J. Geriatr. Psychiatry 2006, 21, 17–28. [DOI] [PubMed] [Google Scholar]
- Çokuğraş A. N. Butyrylcholinesterase: Structure and physiological importance. Turk. J. Biochem. 2003, 28, 54–61. [Google Scholar]
- Giacobini E. Cholinergic function and Alzheimer's disease. Int. J. Geriatr. Psychiatry 2003, 18Suppl. 1S1–S5. [DOI] [PubMed] [Google Scholar]
- Giacobini E.Drugs that target cholinesterases. In Cognitive Enhancing Drugs; Buccafusco J. J., Ed.; Birkhäuser: Basel, Boston, Berlin, 2004; pp 11–36. [Google Scholar]
- Greig N. H.; Utsuki T.; Ingram D. K.; Wang Y.; Pepeu G.; Scali C.; Yu Q. S.; Mamczarz J.; Holloway H. W.; Giordano T.; Chen D.; Furukawa K.; Sambamurti K.; Brossi A.; Lahiri D. K. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning, and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 17213–17218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M. M.; Guillozet A.; Shaw P.; Levey A.; Duysen E. G.; Lockridge O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [DOI] [PubMed] [Google Scholar]
- Carolan C. G.; Dillon G. P.; Khan D.; Ryder S. A.; Gaynor J. M.; Reidy S.; Marquez J. F.; Jones M.; Holland V.; Gilmer J. F. Isosorbide-2-benzyl carbamate-5-salicylate, a peripheral anionic site binding subnanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 2010, 53, 1190–1199. [DOI] [PubMed] [Google Scholar]
- Decker M. Novel inhibitors of acetyl- and butyrylcholinesterase derived from the alkaloids dehydroevodiamine and rutaecarpine. Eur. J. Med. Chem. 2005, 40, 305–313. [DOI] [PubMed] [Google Scholar]
- Decker M.; Krauth F.; Lehmann J. Novel tricyclic quinazolinimines and related tetracyclic nitrogen bridgehead compounds as cholinesterase inhibitors with selectivity towards butyrylcholinesterase. Bioorg. Med. Chem. 2006, 14, 1966–1977. [DOI] [PubMed] [Google Scholar]
- Decker M. Hybrid molecules incorporating natural products: applications in cancer therapy, neurodegenerative disorders and beyond. Curr. Med. Chem. 2011, 18, 1464–1475. [DOI] [PubMed] [Google Scholar]
- Tumiatti V.; Minarini A.; Bolognesi M. L.; Milelli A.; Rosini M.; Melchiorre C. Tacrine derivatives and Alzheimer's disease. Curr. Med. Chem. 2010, 17, 1825–1838. [DOI] [PubMed] [Google Scholar]
- Decker M. Homobivalent quinazolinimines as novel nanomolar inhibitors of cholinesterases with dirigible selectivity toward butyrylcholinesterase. J. Med. Chem. 2006, 49, 5411–5413. [DOI] [PubMed] [Google Scholar]
- Decker M.; Kraus B.; Heilmann J. Design, synthesis and pharmacological evaluation of hybrid molecules out of quinazolinimines and lipoic acid lead to highly potent and selective butyrylcholinesterase inhibitors with antioxidant properties. Bioorg. Med. Chem. 2008, 16, 4252–4261. [DOI] [PubMed] [Google Scholar]
- Montiel T.; Quiroz-Baez R.; Massieu L.; Arias C. Role of oxidative stress on β-amyloid neurotoxicity elicited during impairment of energy in the hippocampus: Protection by antioxidants. Exp. Neurol. 2006, 200, 496–508. [DOI] [PubMed] [Google Scholar]
- Palmer A. M. Neuroprotective therapeutics for Alzheimer's disease: Progress and prospects. Trends Pharmacol. Sci. 2011, 32, 141–147. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zenger K.; Lupp A.; Kling B.; Heilmann J.; Fleck C.; Kraus B.; Decker M. Tacrine-silibinin codrug shows neuro- and hepatoprotective effects in vitro and pro-cognitive and hepatoprotective effects in vivo. J. Med. Chem. 2012, 55, 5231–5242. [DOI] [PubMed] [Google Scholar]
- Fang L.; Kraus B.; Lehmann J.; Heilmann J.; Zhang Y.; Decker M. Design and synthesis of tacrine-ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 2905–2909. [DOI] [PubMed] [Google Scholar]
- Williams P.; Sorribas A.; Howes M. R. Natural products as a source of Alzheimer's drug leads. Nat. Prod. Rep. 2011, 28, 48–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macabeo A. P. G.; Vidar W. S.; Chen X.; Decker M.; Heilmann J.; Wan B.; Franzblau S. G.; Galvez E. V.; Aguinaldo M. A. M.; Cordell G. A. Mycobacterium tuberculosis and cholinesterase inhibitors from Voacanga globosa. Eur. J. Med. Chem. 2011, 44, 1702–1709. [DOI] [PubMed] [Google Scholar]
- Hostettmann K.; Borloz A.; Urbain A.; Marston A. Natural product inhibitors of acetylcholinesterase. Curr. Org. Chem. 2006, 10, 825–847. [Google Scholar]
- Weinstock M.; Groner E. Rational design of a drug for Alzheimer's disease with cholinesterase inhibitory and neuroprotective activity. Chem.-Biol. Interact. 2008, 175, 216–222. [DOI] [PubMed] [Google Scholar]
- Murphy T. H.; Miyamoto M.; Sastre A.; Schnaar R. L.; Coyle J. T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [DOI] [PubMed] [Google Scholar]
- Tan S.; Wood M.; Maher P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J. Neurochem. 1998, 71, 95–105. [DOI] [PubMed] [Google Scholar]
- Tan S.; Schubert D.; Maher P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506. [DOI] [PubMed] [Google Scholar]
- Ishige K.; Schubert D.; Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radical Biol. Med. 2001, 30, 433–446. [DOI] [PubMed] [Google Scholar]
- Lee K. Y.; Hwang L.; Jeong E. J.; Kim S. H.; Kim Y. C.; Sung S. H. Effect of neuroprotective flavonoids of Agrimonia eupatoria on glutamate-induced oxidative injury to HT22 hippocampal cells. Biosci., Biotechnol., Biochem. 2010, 74, 1704–1706. [DOI] [PubMed] [Google Scholar]
- Luo W.; Yu Q.-s.; Zhan M.; Parrish D.; Deschamps J. R.; Kulkarni S. S.; Holloway H. W.; Alley G. M.; Lahiri D. K.; Brossi A.; Greig N. H. Novel anticholinesterases based on the molecular skeletons of furobenzofuran and methanobenzodioxepine. J. Med. Chem. 2005, 48, 986–994. [DOI] [PubMed] [Google Scholar]
- Bolognesi M. L.; Bartolini M.; Cavalli A.; Andrisano V.; Rosini M.; Minarini A.; Melchiorre C. Design, synthesis, and biological evaluation of conformationally restricted rivastigmine analogues. J. Med. Chem. 2004, 47, 5945–5952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.