

Abstract

A series of 1-phenyl-3-(1-phenylethyl)urea derivatives were identified as novel and potent complement inhibitors through structural modification of the original compound from high-throughput screening. Various analogues (7 and 13–15) were synthesized and identified as complement inhibitors, with the introduction of a five- or six-carbon chain (7c, 7d, 7k, 7l, and 7o) greatly improving their activity. Optimized compound 7l has an excellent inhibition activity with IC50 values as low as 13 nM. We demonstrated that the compound 7l inhibited C9 deposition through the classical, the lectin, and the alternative pathways but had no influence on C3 and C4 depositions.
Keywords: high-throughput screening, structural modification, complement inhibitors, C9
The complement system plays an important role in protecting the body against the intrusion of cellular and viral pathogens, as well as in inflammation.1 There are at least 25 complement proteins, which are a complex collection of plasma proteins and membrane cofactors. Complement is activated by three pathways: the classical pathway, the alternative pathway, and the lectin pathway. Under physiological conditions, complement activation is regulated by a series of membrane-bound and soluble complement inhibitors, involved in protecting the body against excessive complement activation. Accumulating data suggest that inappropriate activation of complement is involved in the pathogenesis of many inflammatory, autoimmune, neurodegenerative, and infectious diseases.2,3 Moreover, with the development of improved animal models and genome-wide association studies, it has become apparent that some common and often untreatable diseases such as age-related macular degeneration are largely due to mutations in the complement system. The successful use of Soliris (Eculizumab), a monoclonal antibody specific for C5, and other complement inhibitors indicates that targeting complement is a promising strategy in many clinical situations.4,5
In recent years, several organic small molecule inhibitors of complement have been discovered.6−8 In contrast to protein inhibitors, low molecular weight drugs are cheap and have oral bioavailability and pharmacokinetics advantages. However, most of these inhibitors have proved to be plagued by a variety of problems, including poor selectivity, high toxicity, low potency, and short half-life. There is an urgent need for the development of organic small molecule complement inhibitors for effectively treating complement-related diseases. In this paper, we report 1-phenyl-3-(1-phenylethyl) urea derivatives as novel and potent inhibitors of the complement system through structural modification based on screening hit 1 (Figure 1).
Figure 1.

High-throughput screening hit 1.
Through high-throughput screening (HTS) in the fetal complement hemolytic assay, 1-(3,4-dimethoxyphenyl)-3-(1-phenylethyl)urea (hit 1) was discovered as a hit compound that showed a moderate complement inhibition activity, with a half-maximal inhibitory concentration (IC50) value of 50 μM for the hemolytic assay. This prompted us to start a chemistry program to further explore this class of compounds. In this communication, we would like to report a series of potent complement inhibitors discovered through this effort.
We initially prepared compounds 2a–d (Figure 2), with modification of the right-hand side of compound 1, in which the S-phenylethylamino group in compound 1 was replaced with biphenylmethylamino (2a), naphthylmethylamino (2b), R-phenylethylamino (2c), and N-methylbenzylamino (2d) groups. Compounds 2a–d were synthesized according to Scheme 1. Dimethoxybenzoic acid 3 was converted to the corresponding aryl isocyanate 4 with diphenyl phosphorazidate (DPPA) and triethylamine (Et3N) via a modified Curtius rearrangement,9−9c followed by treatment of isocyanate 4 with the corresponding amines to afford the desired compounds 2a–d.
Figure 2.

Compounds 2a–d.
Scheme 1. Synthesis of Compounds 2a–d.

Reagents and conditions: (a) DPPA, Et3N, benzene, then reflux. (b) DCM, 45–80% (two steps).
Among these ureas, compounds 2a and 2b did not show any complement inhibition at 100 μg/mL concentration, while compounds 2c and 2d showed 16.95 and 43.8% inhibition at 100 μg/mL, respectively. These initial attempts indicated that the phenylethyl subunit in hit 1 is essential for effective inhibition of complement activation. It is worth mentioning that compound 2c, a R-enantiomer of compound 1, showed very weak inhibition (16.95% inhibition at 100 μg/mL) as compared to compound 1 (IC50 = 50 μM) in the hemolytic assay. These results indicated that the S-configuration of the chiral center in this position is very critical.
Then, we switched our focus to the left-hand side benzene ring subunit of compound 1. Five compounds (5a–e) were prepared (Table 1), in which the dimethoxyphenyl in compound 1 was replaced with various substituted phenyls and pyridine. The syntheses of compounds 5a–e in Table 1 were similar to compounds 2a–d. Substituted benzoic acid was converted to the corresponding aryl isocyanate. Treatment of the isocyanate with 1-phenylethylamine led to the desired compounds 5a–e.
Table 1. Compounds 5a–e.
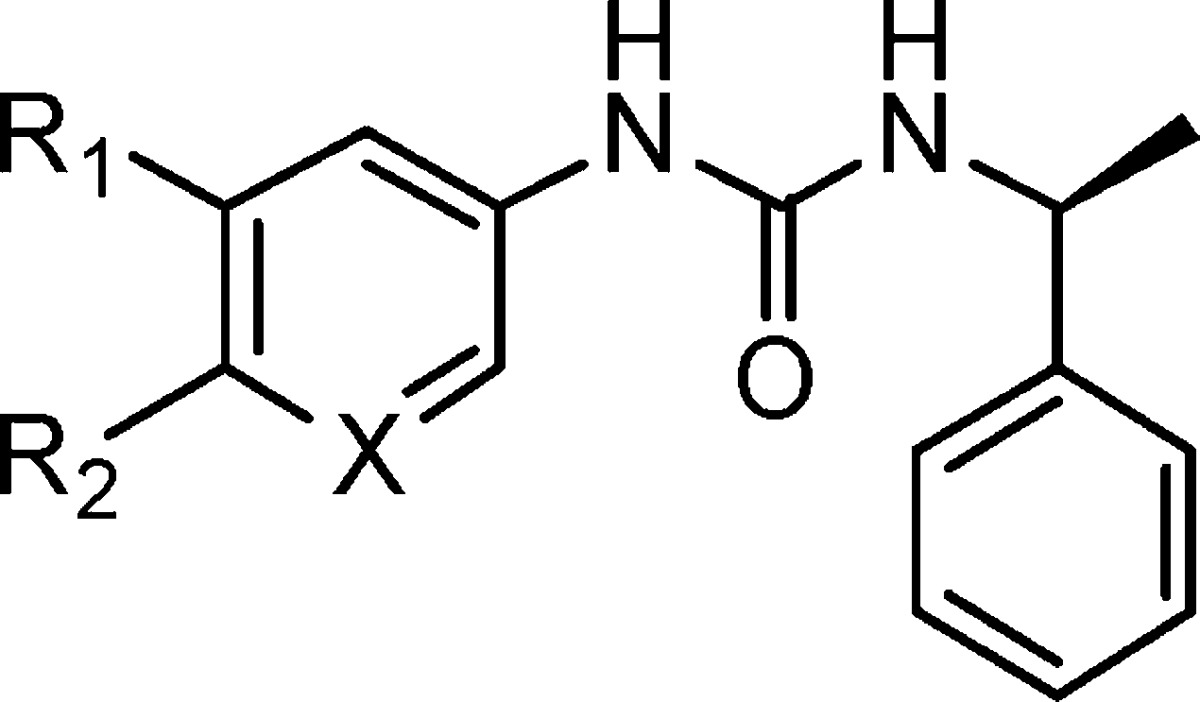
| compd | R1 | R2 | X |
|---|---|---|---|
| 5a | H | H | C |
| 5b | H | Cl | C |
| 5c | H | OMe | C |
| 5d | OMe | H | C |
| 5e | H | H | N |
None of these compounds showed any activity against complement in hemolytic assay at a concentration of 100 μg/mL. These results indicate that the substituent in the left-hand phenyl group is more sensitive. To understand the contribution of the two methoxyl groups to complement inhibition activity, a focused library was designed by replacement of the methyl with various substituted groups, including H, benzyl, Et, allyl, and carboxylalkyl- and amino-substituted alkyls (Table 2).
Table 2. Compounds 6a–k.
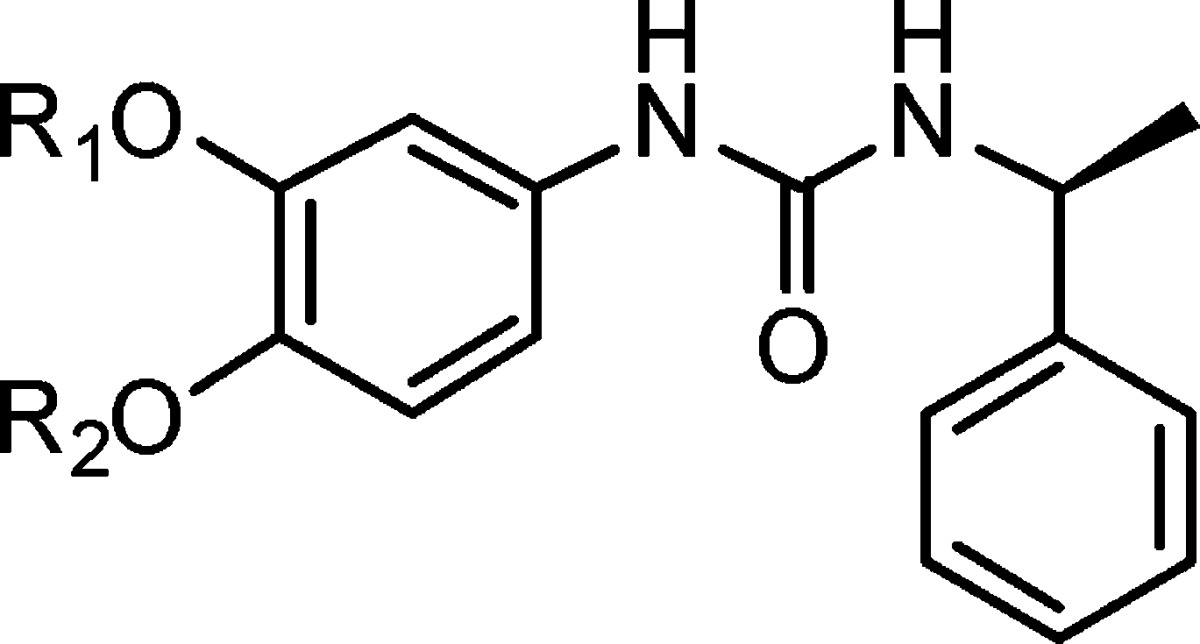
| compd | R1 | R2 | IC50 (μM) |
|---|---|---|---|
| 6a | H | Me | 35.28% inhibition at 100 μg/mL |
| 6b | benzyl | Me | 32.71% inhibition at 100 μg/mL |
| 6c | Et | Me | 2.86 ± 0.38 |
| 6d | allyl | Me | 1.75 ± 0.21 |
| 6e | EtO2CCH2– | Me | 6.20 ± 0.31 |
| 6f | Me | H | 14.62% inhibition at 100 μg/mL |
| 6g | Me | Benzyl | NAa |
| 6h | Me | Et | NA |
| 6i | Me | Allyl | NA |
| 6j | Me | EtO2CCH2– | NA |
| 6k | Me | BocNHCH2CH2– | NA |
Showed no inhibition at 100 μg/mL concentration.
The inhibitory activities of these compounds are summarized in Table 2. The activity of these compounds varied greatly. 3-Position methyl substituted with Et (6c), allyl (6d), or ethyl acetate (6e) resulted in ca. 9–28-fold improvement of activity as compared with compound 1. Substitution with other substituents such as benzyl (6b) or H (6a) resulted in weaker activity as compared with compound 1. 4-Position methyl substituted with benzyl, Et, allyl, or other groups all led to inactive compounds (6f–k). 3-Position methyl replacements were tolerated. The above results also indicate that the size of the substituent in the 3-position has a great impact on the activity and therefore provides a suitable point for further modification to improve the activity.
A follow up-focused library (outlined in Scheme 2) changing the methyl for a series of 3-alkyl groups was then synthesized. In this library, we kept both the 4-methoxy in the left-hand side benzene ring and the S-phenylethyl in the right-hand side.
Scheme 2. Synthesis of Compounds 7a–x.

Reagents and conditions: (a)Benzyl bromide, K2CO3, acetone, 98%. (b) 30% H2O2, NaH2PO4, NaClO2, acetonitrile, H2O, 72%. (c) DPPA, Et3N, benzene, then reflux. (d) (S)-α-Phenylethylamine, DCM, 48% (two steps). (e) Hydrogen, 5% palladium on activated carbon, DMF, 85%. (f) Alkyl bromide, K2CO3, 18-crown-6, DMF, 80–95%.
The syntheses of compounds 7a–x in Table 3 were similar to those of compounds 2a–d in Scheme 2. Benzylation of starting material isovanillin 8 was achieved to benzaldehyde 9, which was then oxidized to carboxylic acid 10 applying a known method.10 Carboxylic acid 10 was converted to isocyanate 11. Treatment of the isocyanate with (S)-α-phenylethylamine yielded compound 6b. Hydrogenolytic cleavage of the benzyl ether group of compound 6b using H2/Pd/C provided compound 6a. Treatment of compound 6a with various alkyl bromides in the presence of K2CO3 provided corresponding compounds 7a–x.
Table 3. Compounds 7a–x.
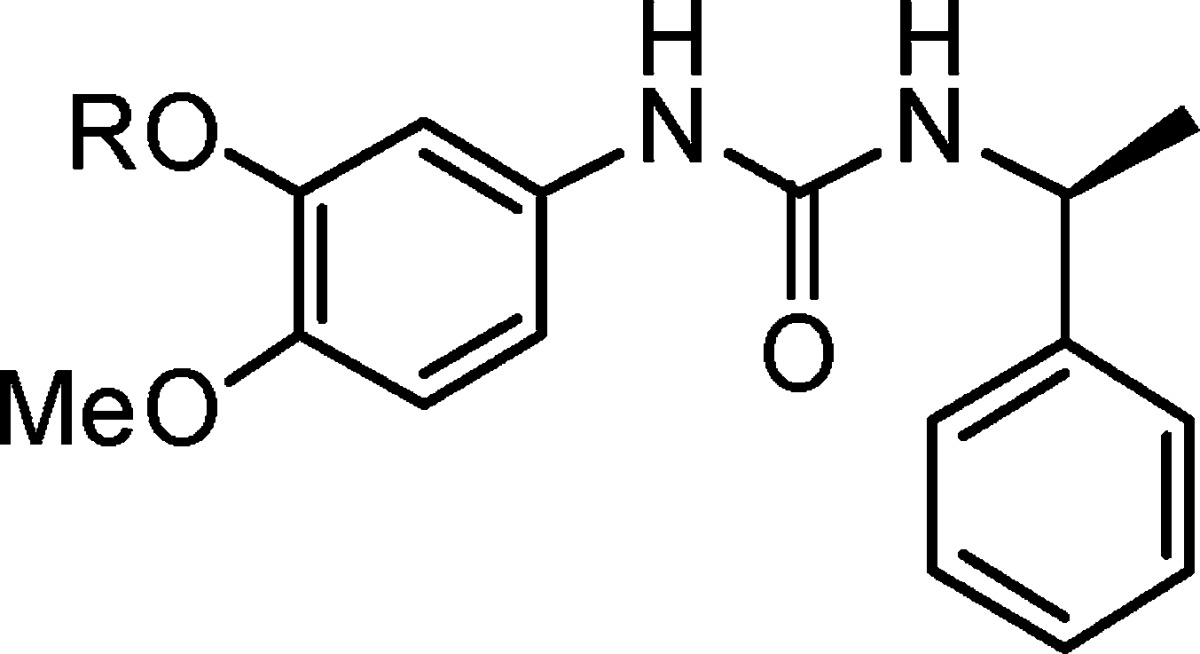

Showed no inhibition at 100 μg/mL concentration.
The inhibitory activities of these compounds toward complement are summarized in Table 3. Most of this series of compounds showed potent inhibition activity, with the trends of the length of side chain significantly affecting the activity and heteroatom introduction in the side chain removing the activity. Among 24 compounds tested (7a–7x), 21 of them were more active than the lead molecule 1. In particular, compounds with a five- or six-carbon chain (7c, 7d, 7k, 7l, and 7o) showed a dramatic increase of up to 1000-fold improvement of activity as compared with compound 1. Longer side chains (7g, 7h) resulted in no inhibition because of poor solubility. The overall structure–activity relationship (SAR) is alkane > alkyl > alkynyl > cycloalkane > heterocycloalkane. The compound BCX1470 was used as the positive control. It is a potent inhibitor of C1s and factor D in vitro, and it prevents the initial phase of IC-mediated inflammation and associated systemic complement activation in rats.11
As a further extension of the SAR on the substituent positions on the left-hand side benzene ring, we synthesized several compounds, 12a–f, with different substituted positions of the methoxyl of C5 alkyloxyl group, as outlined in Figure 3.
Figure 3.

Compounds 12a–f.
Compounds 12c–f showed no inhibitory activity against complement. Compounds 12a and 12b exhibited an IC50 of 3.2 and 0.27 μM in hemolytic assays, respectively, which were all weaker than those of the closely related compound 7c (IC50 = 0.019 μM). These results, combined with the data from the series of 6c and 7a–r, indicated that the 4-methoxyl group, as well as an appropriate size alkyl substituent in the 3-position, is essential for effective inhibition of complement. Hydrophobic interaction of 3- and 4- position groups with target proteins might be responsible for an increase in potency, which may be worth further investigation for confirmation.
Having synthesized the potent inhibitor 7b, we then moved our attention to the right-hand side modification for further improvement of potency. Compounds 13a and 13b were designed to examine the length between the phenylethyl and the N atom, while 13c, 13d, 13e, and 13f were designed to determine the contribution of the configuration and restriction or extension of the key methyl group to inhibitory activity (Figure 4).
Figure 4.

Compounds 13a–f.
Compounds 13a–d showed no activity. Compound 13e exhibited an IC50 of 0.105 μM, and compound 13f was 0.42 μM. The two compounds were all weaker than that of the closely related compound 7b (IC50 = 0.033 μM). The results also indicate that the size of the connection had a great impact on inhibition activity. We also synthesized compounds 14a–g, in which the N atom of the amide bond was alkylated with different size groups (Table 4).
Table 4. Compounds 14a–h.
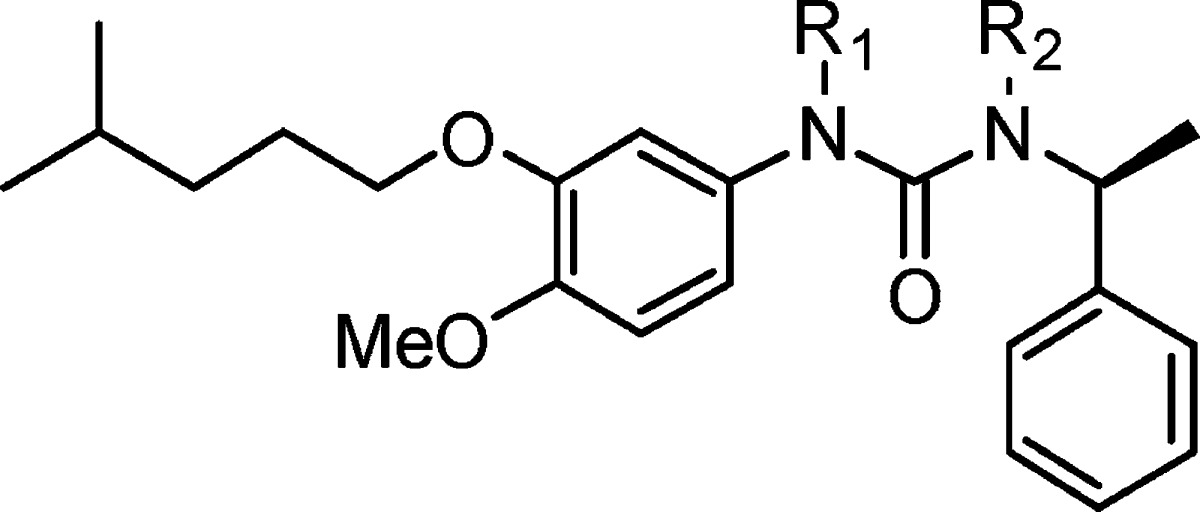
| compd | R1 | R2 | IC50 (μM) |
|---|---|---|---|
| 7l | H | H | 0.013 ± 0.001 |
| 14a | H | Me | 0.019 ± 0.047 |
| 14b | Me | H | 0.54 ± 0.123 |
| 14c | Me | Me | NAa |
| 14d | H | CH2CH2OH | 0.323 ± 0.026 |
| 14e | H | CH2CHOHCH2OH | 0.368 ± 0.087 |
| 14f | CH2CH2CH2OH | H | 1.58 ± 0.307 |
| 14g | H | CH2CH2CH2OH | 0.223 ± 0.047 |
| 14h | H | CH2CH2CH2NH2 | 7.16 ± 0.02 |
Showed no inhibition at 100 μg/mL concentration.
Except for compound 14a with a methyl group on the right-hand side N atom, whose activity (IC50 = 0.019 μM) was similar to compound 7l (IC50 = 0.013 μM), the other compounds exhibit 10–200-fold decrease in activity as compared with compound 7l. This indicates that the free left-hand side NH is necessary to keep the high potency of inhibition activity, while the right-hand side NH tolerates small size substitutions.
We further studied the right-hand side substituent on the benzene ring by keeping all of the favored substituted groups, including 3-isohexyl, 4-methyl, two free NH as well as S-configuration methylphenylamine and with various substitutions on the benzyl ring. Among the synthesized compounds 15a–l (Table 5), a compound with a substituted hydroxyl exhibited better activity (15f), while compounds with 3-bromine, 4-fluorine, or methyl substitutions resulted in ca. 30–100-fold decrease in activity. Overall, the SAR in this position is flat, almost of these modifications resulted in similar or weaker activities as compared with that of 7l, which has an unsubstituted phenyl group. Compounds with electron-donating groups exhibited better activity, while compounds with electron-withdrawing groups resulted in ca. 30–100-fold decrease in activity. Further studies are needed to clarify the role of SAR.
Table 5. Compounds 15a–l.
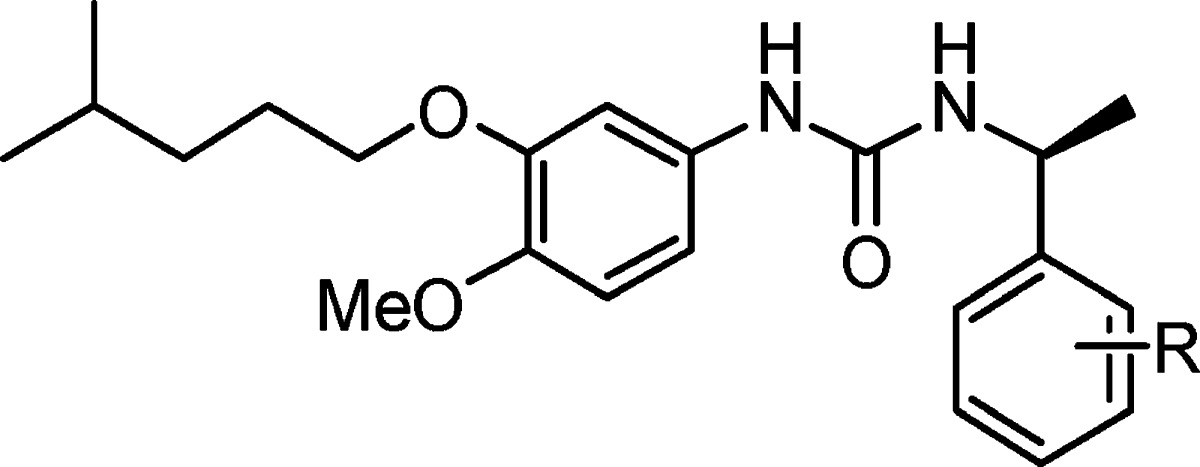
| compd | R | IC50 (μM) |
|---|---|---|
| 7l | H | 0.013 ± 0.001 |
| 15a | 3-Br | 1.32 ± 0.083 |
| 15b | 4-Br | 3.39 ± 0.173 |
| 15c | 2-NO2 | 0.368 ± 0.059 |
| 15d | 4-NO2 | 1.69 ± 0.48 |
| 15e | 4-OH | 0.139 ± 0.029 |
| 15f | 3-OH | 0.077 ± 0.009 |
| 15g | 4-F | 0.402 ± 0.038 |
| 15h | 4-CF3 | NAa |
| 15i | 4-CN | 1.26 ± 0.059 |
| 15j | 4-CO2Me | 0.535 ± 0.051 |
| 15k | 4-CO2H | 0.761 ± 0.178 |
| 15l | 4-Me | 0.327 ± 0.040 |
Showed no inhibition at 10 μg/mL concentration.
Compound 7l was selected for studies delineating the mechanism of inhibitory action on complement. The results indicated that compound 7l did not interfere with the C4 and C3 activation but inhibited the membrane attack complex formation (C9 deposition) by the three pathways. The IC50 was approximately 3.68, 4.46, and 3.59 μg/mL for the classical, lectin, and alternative pathways, respectively. There was almost 90% inhibition at 10 μg/mL (Figure 5).
Figure 5.

Compound 7l inhibited C9 deposition through the classical, the lectin, and the alternative pathways but had no influence on C3 and C4 depositions. Normal human serum (NHS) was added to IgM-coated plates (classical pathway), mannan-coated plates (lectin pathway), or zymosan-coated plates (alternative pathway) and then incubated with various concentrations of compound 7l. Deposited C3 (A), C4 (B), and C9 (C) were measured by ELISA.
In summary, we have discovered a novel series of complement inhibitors based on 1-(3,4-dimethoxyphenyl)-3-(1-phenylethyl)urea. Extensive SAR studies were carried out on both the left-hand dimethoxyphenyl moiety and the right-hand phenylethylamine moiety of the initial HTS lead compound 1, which resulted in the identification of potent inhibitors with IC50 values as low as 13 nM (7l). We also demonstrated that the compound 7l inhibited C9 deposition but had no influence on the preceding C3 and C4 deposition by the classical, the lectin, and the alternative pathway. These compounds could serve as leads for the development of small molecule agents targeting the complement system.
Glossary
Abbreviations
- DCM
dichloromethane
- DPPA
diphenylphosphoryl azide
- Et3N
triethylamine
- HTS
high-throughput screening
- NHS
normal human serum
Supporting Information Available
Experimental procedures and characterization of new chemical entities. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. These authors contributed equally.
This work was supported by The National Natural Science Foundation of China (Grants 30725049, 81021062, and 81072652) and by the grant of Shanghai Science and Technology Committee (No. 11431921102).
The authors declare no competing financial interest.
Supplementary Material
References
- Barrington R.; Zhang M.; Fischer M.; Carroll M. C. The role of complement in inflammation and adaptive immunity. Immunol. Rev. 2001, 180, 5–15. [DOI] [PubMed] [Google Scholar]
- Kirschfink M. Targeting complement in therapy. Immunol. Rev. 2001, 180, 177–189. [DOI] [PubMed] [Google Scholar]
- Caliezi C.; Wuillemin W. A.; Zeerleder S.; Redondo M.; Eisele B.; Hack C. E. C1-esterase inhibitor: An anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharmacol. Rev. 2000, 52, 91–112. [PubMed] [Google Scholar]
- Wagner E.; Frank M. M. Therapeutic potential of complement modulation. Nat. Rev. Drug Discovery 2010, 9, 43–56. [DOI] [PubMed] [Google Scholar]
- Ricklin D.; Lambris J. D. Complement-targeted therapeutics. Nat. Biotechnol. 2007, 25, 1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland M. C. H.; Morikis D.; Lambris J. D. Synthetic small molecule complement inhibitors. Curr. Opin. Invest. Drugs 2004, 5111164–1173. [PubMed] [Google Scholar]
- Kozlov L. V.; Burdelev O. O.; Bureeva S. V.; Kaplun. A. P. Artificial inhibition of the complement system. Russ. J. Bioorg. Chem. 2007, 335449–473. [DOI] [PubMed] [Google Scholar]
- Qu H.; Ricklin D.; Lambris J. D. Recent developments in low molecular weight complement inhibitors. Mol. Immunol. 2009, 47, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioiri T.; Ninomiya K.; Yamada S.-i. Diphenylphosphoryl azide. New convenient reagent for a modified Curtius reaction and for peptide synthesis. J. Am. Chem. Soc. 1972, 94, 6203–6205. [DOI] [PubMed] [Google Scholar]
- Ninomiya K.; Shioiri T.; Yamada S. Phosphorus in organic synthesis—VII: Diphenyl phosphorazidate (DPPA). A new convenient reagent for a modified curtius reaction. Tetrahedron 1974, 30, 2151–2157. [Google Scholar]
- Rigby J. H.; Balasubramanian N. Preparation of highly substituted 2-pyridones by reaction of vinyl isocyanates and enamines. J. Org. Chem. 1989, 54, 224–228. [Google Scholar]
- Dalcanale E.; Montanari F. Selective oxidation of aldehydes to carboxylic acids with sodium chlorite-hydrogen peroxide. J. Org. Chem. 1986, 51, 567–569. [Google Scholar]
- Szalai A. J.; Digerness S. B.; Agrawal A.; Kearney J. F.; Bucy R. P.; Niwas S.; Kilpatrick J. M.; Babu Y. S.; Volanakis J. E. The Arthus reaction in rodents: Species-specific requirement of complement. J. Immunol. 2000, 164, 463–468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.