

Abstract

This letter provides the first pharmacological proof of principle that the sst3 receptor mediates glucose-stimulated insulin secretion (GSIS) from pancreatic β-cells. To enable these studies, we identified the selective sst3 antagonist (1R,3R)-3-(5-phenyl-1H-imidazol-2-yl)-1-(tetrahydro-2H-pyran-4-yl)-2,3,4,9-tetrahydro-1H-β-carboline (5a), with improved ion channel selectivity and mouse pharmacokinetic properties as compared to previously described tetrahydro-β-carboline imidazole sst3 antagonists. We demonstrated that compound 5a enhances GSIS in pancreatic β-cells and blocks glucose excursion induced by dextrose challenge in ipGTT and OGTT models in mice. Finally, we provided strong evidence that these effects are mechanism-based in an ipGTT study, showing reduction of glucose excursion in wild-type but not sst3 knockout mice. Thus, we have shown that antagonism of sst3 represents a new mechanism with potential in treating type 2 diabetes mellitus.
Keywords: somatostatin, type 2 diabetes, sst3, glucose-stimulated insulin secretion
Type 2 diabetes mellitus (T2DM) accounts for 90–95% of clinical diabetes and represents a major and growing health threat throughout the world. The overall estimated worldwide prevalence of diabetes in 2000 was 2.8% (171 million people), with that number projected to increase to 4.4% (366 million people) by 2030.1 Within the United States, a 2001 estimate of the prevalence of diagnosed diabetes cases was even greater at 7.9% of the population (16.7 million people), with an even larger number of sufferers believed to be undiagnosed.1 Because of the trend of increasing rates of obesity in the United States and developing world, the vast majority of newly diagnosed cases of diabetes are anticipated to fall within T2DM spectrum.2 Diabetes mellitus carries a tremendous human suffering and economic burden as a consequence of the associated chronic complications (blindness, end-stage renal disease, limb amputations, heart disease, stroke, etc.) and increased morbidity and mortality. Unfortunately, despite numerous medication options for the treatment of T2DM, most patients fail to achieve long-term glycemic control, and new treatment options are urgently needed.3
A decline in glucose-stimulated insulin secretion (GSIS) from pancreatic β-cells is one of the primary defects associated with the development and progression of T2DM. Agents such as sulfonyl ureas and related K-ATP channel blockers that increase insulin secretion from β-cells are efficacious in lowering glucose, but because their effects are continuous and not dependent on plasma glucose levels, they carry the risk of evoking hypoglycemia and accelerating the loss of islet function. As such, identification of novel targets for stimulating GSIS has been a major strategy in the quest for new agents to treat T2DM;4,5 DPP-4 inhibitors such as sitagliptin represent one recently introduced class of such agents. As part of our continuing efforts to identify novel GSIS targets, Zhou and co-workers recently determined that the somatostatin receptor subtype 3 (sst3) is highly expressed in β-cells of human and rodent islets, and knocking down expression of sst3 using siRNA technology enhances GSIS in INS-1 cells (a rat insulinoma cell line).6 Somatostatin (somatotropin release-inhibiting factor, SRIF) is a cyclic tetradecapeptide existing in two biologically active forms, SRIF-14 and N-terminal extended SRIF-28, that acts as a neurotransmitter and hormone with broad inhibitory effects on both endocrine (growth hormone, insulin, glucagon, gastrin, cholecystokinin, etc.) and exocrine (gastric acid, pancreatic enzymes, etc.) secretion. These effects are mediated through interactions with five G-protein-coupled somatostatin receptor subtypes (sst1, sst2, sst3, sst4, and sst5).7 Of the five somatostatin receptor subtypes, the specific physiological functions of the individual receptor subtypes are best understood for sst2 and sst5.8 In contrast to sst2 and sst5, relatively little is known about the function of sst3. Expression of sst3 has been described in brain, pituitary, stomach, pancreas, thymus, thyroid, prostate, vascular endothelial cells, and various human tumors. The sst3 receptor has been linked to apoptotic signaling and proliferation and has been proposed as a possible target for cancer therapy.9−11 To our knowledge, a role for the sst3 receptor in regulating GSIS of pancreatic β-cells had not been previously described prior to this report. This letter describes the development of an sst3 antagonist with suitable somatostatin receptor subtype and off target selectivity and rodent pharmacokinetic properties, to provide in vivo proof of principal that antagonism of sst3 represents a viable novel strategy for stimulation of GSIS and, possibly, for treatment of T2DM.
Relatively few small molecule nonpeptide sst3 antagonists have been described in the literature.12 Several examples are shown in Figure 1. Compound 1 was described by Novartis scientists as a potent, selective silent competitive sst3 antagonist (human sst3 binding affinity <10 nM, selectivity against human sst1, sst2, sst5 all >150-fold; selectivity against sst4 21-fold; cAMP functional antagonist potency pKB = 7.88).13,14 Tetrahydro-β-carboline imidazole compounds 2 and 3 were described by Poitout, Thurieau, and co-workers at the Institut Henri Beaufour as having high binding affinities for sst3 (0.64 and 1.7 nM, respectively), both with >1000-fold selectivity over the other somatostatin receptor subtypes.15 In functional assays, these compounds behaved as competitive antagonists of sst3. Novartis scientists described closely related β-carboline imidazole sst3 antagonists such as 4 (sst3 human pKd 8.69, >400-fold selective against other receptor subtypes) in a recently issued patent, which claims these compounds as potential treatments for depression, anxiety, bipolar disorder, and cancer.16 Unfortunately, in our hands, compounds 2–4 had poor pharmacokinetic properties in mice with high clearance rates (126, 95, and 31 mL/min/kg, respectively) and poor plasma exposures following oral dosing. This limited the usefulness of these compounds as tools for in vivo pharmacological assessment of sst3 as a potential new GSIS target. Moreover, while not reported for these compounds, based on internal assays, compounds 2–4 had high affinities for several cardiac ion channels including the hERG channel (binding IC50 = 150, 77, and 176 nM, respectively) and/or the L type Ca2+ channel (binding IC50 = 128, 308, and 1526 nM, respectively). We chose to explore modifications of compounds related to 2–4 in an effort to identify novel analogs with improved pharmacokinetic and off-target profiles. On the basis of the slight improvement in Ca2+ channel selectivity observed upon incorporation of oxygen atoms into the n-butyl groups of 2 leading to 4, we decided to combine features of compounds 3 and 4 by making 1-heterocycle substituted β-carboline analogues (5), a significant divergence from the original structure of the series reported in Novartis and Institut Henri Beaufour patents.
Figure 1.

Previously described sst3 selective antagonists.
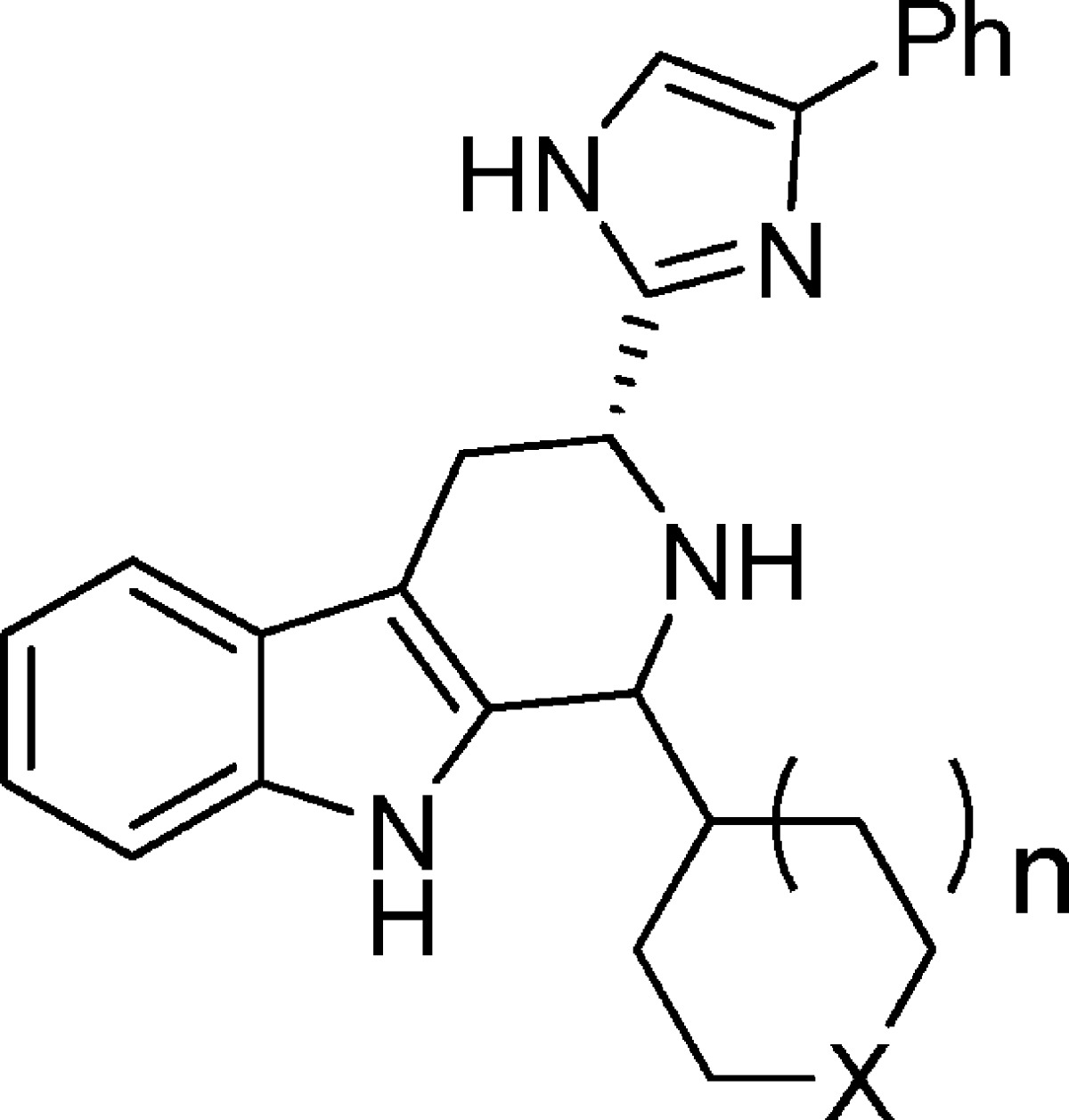
Analogues were readily prepared according to Scheme 1. Boc-d-Trp was treated with bromoacetophenone in the presence of cesium carbonate to provide ester 6. The addition of toluene and ammonium acetate and removal of water via Dean–Stark trap at reflux afforded imidazole 7. Removal of the Boc group (TFA) was followed by Pictet–Spengler cyclization with commercially available or routinely prepared aldehydes. Cyclization generally led to predominantly one isomeric product. Poitout and Thurieau observed the same for compound 3 and assumed that the major isomer had the trans stereochemistry; however, they did not offer any evidence to support this assignment.15 On the basis of a strong NOE in the 1D NOE NMR spectrum between the two methine protons in major isomer 5a, we have established that the relative stereochemistry must be cis instead. A second NOE study with a related analogue (not shown) also demonstrated that the major isomer is cis, giving us added confidence that, for these analogues, the Pictet–Spengler reaction produces predominantly the cis isomers. In some cases, further elaboration was performed under routine conditions (for example, when the 1-substituent was Boc-piperidine, the Boc was removed, and further acylation or sulfonylation could be performed).
Scheme 1. Synthesis of β-Carboline Analogues 5.

Compounds 5a–n were evaluated in a sst3 binding assay,17 a cAMP assay measuring sst3 functional antagonism potency against SS-14,18 and a hERG binding assay.19 The resulting IC50 values for each assay are displayed in Table 1. Replacement of the cyclohexyl moiety in 3 with a variety of saturated heterocycles led, in general, to loss of binding potency (with the exception of compound 5e, which had similar binding potency). Despite this, a number of analogues retained equivalent or better potency as compared to 3 in the assay measuring functional antagonism activity (analogues 5a, 5c, 5d, 5e, 5g, and 5k). Generally, the directional changes in sst3 binding affinity and functional potencies for each analogue track together, but that is not the case for all molecules. Structural changes among the series may affect ligand–receptor on-rates, off-rates, or both and, as such, can have differential effects on the binding affinities and functional antagonism potencies determined in the two different assay formats, wherein the binding assay is a competition readout at steady-state equilibrium, while the cAMP assay is an accumulation challenge assay following prebinding of the test molecule. Installation of a heterocycle, in some cases, also led to a modest to substantial reduction in binding affinity for the hERG channel (analogs 5a, 5j, 5k, 5l, and 5m), demonstrating the feasibility of improving selectivity over hERG in the β-carboline sst3 antagonist class through incorporation of heteroatoms into this fragment. As indicated above, Pictet–Spengler cyclization led to predominantly one stereoisomer product, which in the case of compound 5a, we have established to be cis. Comparison of the potency of the isomerically pure cis (5a) and trans (5b) isomers indicated that the major cis isomer is more potent in both the sst3 binding and the functional assays. On the basis of the comparable functional potency and 5-fold weaker hERG binding affinity (hERG IC50 = 380 nM) of compound 5a as compared to 3, we decided to further profile it. Compound 5a was found to have good selectivity against the other human somatostatin receptor subtypes in binding assays (sst1, sst2, sst4, and sst5 IC50 values all >1 μM). Because our pharmacodynamic model was established in mice, we evaluated compound 5a in the mouse sst3 binding and cAMP-based functional assays (sst3 binding IC50 = 8.9 nM, sst3 cAMP IC50 = 16 nM), where it demonstrated equivalent potency to that observed on the human receptor. When screened against other mouse somatostatin receptor subtypes, it maintained good sst3 subtype selectivity (IC50 msst1, msst2, msst4, and msst5 all >10 μM). It demonstrated weaker potency as compared to compound 3 in the L type Ca2+ channel binding assay (Ca2+ IC50 = 1.9 μM). In a pharmacokinetic study in mice, compound 5a had a reduced clearance rate, increased half-life, higher oral bioavailability, and significantly higher plasma exposures (po) as compared to parent compounds 3 and 4 (Table 2).
Table 1. Human sst3 Binding and Functional Data and hERG Binding Data for 3 and 5a–na.
| IC50 (nM) |
||||
|---|---|---|---|---|
| analogue (n is 1 except as noted) | X | sst3 binding | sst3 cAMP | hERG bindingb |
| 3 | CH2 | 0.8 (5; SD 0.3) | 26 (2) | 77 (2) |
| 5a (cis) | O | 14 (425; SD 4.6) | 22 (417; SD 17) | 380 (2) |
| 5b (trans) | O | 390 (2) | 1200 (1) | 730 (2) |
| 5c, n is 0 isomer 1 | O | 45 (2) | 19 (1) | 64 (2) |
| 5d, n is 0 isomer 2 | O | 63 (2) | 30 (1) | 25 (2) |
| 5e | S | 1.7 (1) | 10 (1) | 120 (2) |
| 5f isomer 1 | SO | 220 (1) | 380 (2) | |
| 5g isomer 2 | SO | 21 (1) | 24 (2) | 100 (2) |
| 5h | SO2 | 120 (2) | 290 (2) | 100 (2) |
| 5i | NBoc | 7 (2) | 1000 (2) | |
| 5j | NH | 63 (1) | 220 (2) | 5100 (2) |
| 5k | NAc | 16 (2) | 46 (2) | 9800 (1) |
| 5l | NSO2Me | 44 (2) | 3500 (2) | |
| 5m | NCO2Me | 4 (2) | 66 (2) | 700 (2) |
| 5n | NCONHMe | 35 (2) | 180 (2) | 240 (2) |
Numbers in parentheses represent numbers of determinations and standard deviation (SD), when calculable.
hERG binding data were obtained by measuring displacement of 35S-MK499 from HEK-293 cells stably expressing hERG.19
Table 2. Pharmacokinetic Properties of Compounds 3, 4, and 5a in C57BL/6N Micea.
| no. | F % | AUCN po (μM h/mg) | Cmax po (nM) | Vdss (L/kg) | Cl (mL/min/kg) | t1/2 (h) |
|---|---|---|---|---|---|---|
| 3 | 15 | 0.07 | 60 | 8.1 | 95 | 0.14 |
| 4 | 4 | 0.06 | 130 | 0.7 | 31 | 0.54 |
| 5a | 45 | 1.3 | 840 | 1.6 | 15 | 1.6 |
Mouse compound exposures were analyzed in blood; iv and po doses were 1 and 2 mg/kg, respectively.
Compound 5a was used to evaluate the role of sst3 in GSIS from the β-cell (Figure 2). In this assay,17 the effects of 5a at 10 μM, octreotide (octr, a peptide somatostatin agonist at 50 nM), a combination of 5a (10 μM) and octreotide at 50 nM, and negative (DMSO) and positive controls [glucagon-like peptide-1 (GLP-1) at 10 nM] on pancreatic islets isolated from normal C57BL/6J mice were determined in the presence of 2 and 16 mM glucose.20 As shown, 5a significantly enhances GSIS (at 16 mM glucose) relative to DMSO control (44% increase). On the other hand, it did not affect the insulin secretion at basal (2 mM) glucose in this assay. This result provides evidence that the sst3 antagonist only enhances GSIS, not basal insulin secretion. GSIS is decreased significantly (by 39%) in the presence of the somatostatin agonist octreotide; however, 5a (at 10 μM) can partially overcome the effect of 50 nM octreotide on GSIS. GLP-1 serving as the positive control (at 10 nM) elevates GSIS as expected.
Figure 2.

Effect of compound 5a (10 μM) on GSIS in pancreatic β-cells from normal C57BL/6J mice (n = 4).
On the basis of the favorable mouse in vitro sst3 potency, the pharmacokinetic profile, and the effect on GSIS in isolated mouse islets of compound 5a, we next sought to evaluate its ability to improve glucose tolerance in ip and po glucose tolerance test models (ipGTT and OGTT, respectively) in lean C57BL/6N Tac mice fasted for 5–6 h before dextrose challenge.21 Figure 3 shows that dextrose challenge causes blood glucose excursion, which in both studies is ameliorated in a dose-dependent fashion by predosing (1 h prior to challenge) with compound 5a (2 and 10 mg/kg). To demonstrate that these effects are mechanism-based, we evaluated compound 5a in an ipGTT study simultaneously comparing effects in wild-type and sst3 knockout mice. As shown in Figure 4, compound 5a reduced glucose excursion resulting from dextrose challenge in the wild-type mice (26% reduction in total blood glucose AUC) but had no significant effect on glucose levels following dextrose challenge in sst3 knockout mice. Blood insulin levels were not monitored in this and other typical GTT studies to avoid excessive stress to the animals and impacting glucose readout. However, in a parallel study in wild-type and knockout mice predosed at −60 min with 10 mg/kg of 5a, a 50% elevation in plasma insulin levels was observed 10 min postdextrose challenge in wild-type mice with no effect on the knockout mice; however, because of variability, this effect did not reach statistical significance (p = 0.12, n = 5).
Figure 3.
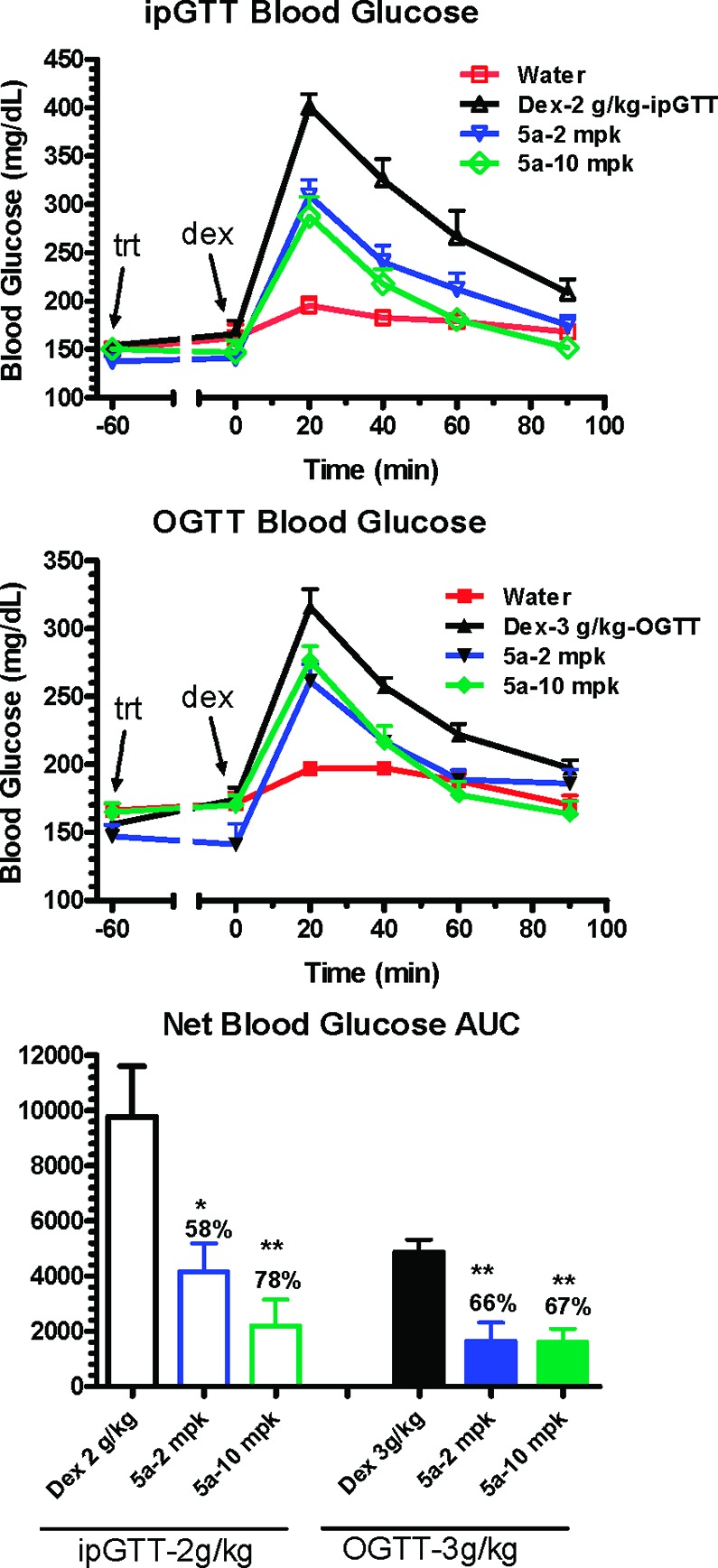
Evaluation of 2 and 10 mg/kg doses of compound 5a in ipGTT and OGTT tests in C57BL/6N Tac mice following dextrose (dex) challenge. *p < 0.05, **p < 0.005 vs dextrose alone. Treatment = trt.
Figure 4.
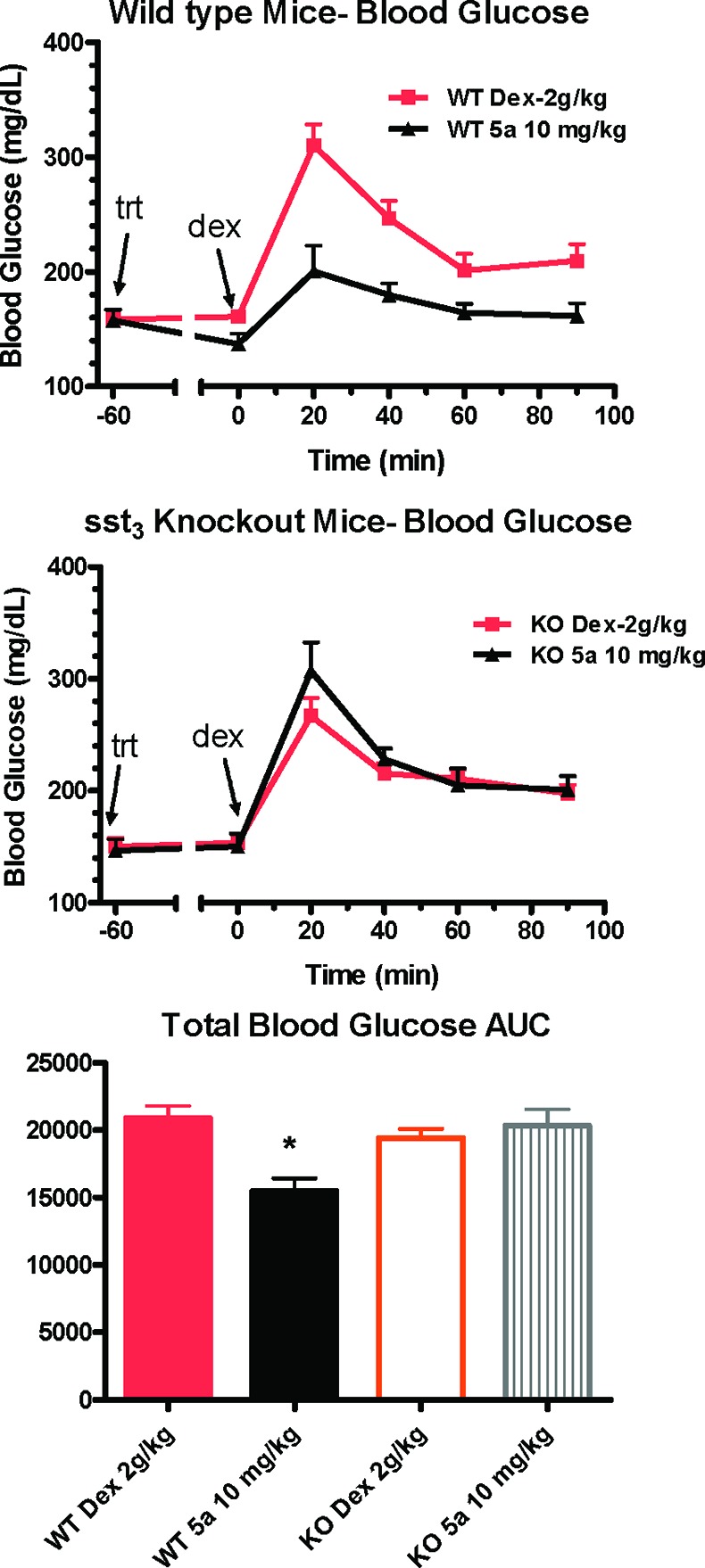
Evaluation of compound 5a (10 mg/kg) in ipGTT studies in wild-type and sst3 knockout mice challenged with 2 g/kg dextrose (dex). *p < 0.05 vs dextrose alone. Treatment = trt.
In summary, we have recently shown for the first time that the sst3 receptor mediates GSIS from pancreatic β-cells. To provide pharmacological proof of principle for this observation, we developed β-carboline imidazole-based selective sst3 antagonist 5a, with improved ion channel selectivity and mouse pharmacokinetic characteristics as compared to previously described structurally related antagonists 2–4. We demonstrated that compound 5a enhances GSIS in pancreatic β-cells and blocks glucose excursion induced by dextrose challenge in ipGTT and OGTT models in mice. Moreover, we provided strong evidence that these effects are mechanism-based in an ipGTT study showing blockade of glucose excursion in wild-type but not sst3 knockout mice. Thus, we have shown that antagonism of sst3 represents a new GSIS target with potential in treating T2DM.
Supporting Information Available
Experimental details for the synthesis and characterization of sst3 antagonist 5a, in vitro assays, GSIS assay with pancreatic islets, and GTTs. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Wild S.; Roglic G.; Green A.; Sicree R.; King H. Global Prevalence of Diabetes: Estimates for the Year 2000 and Projections for 2030. Diabetes Care 2004, 27, 1047–1053. [DOI] [PubMed] [Google Scholar]
- Zimmet P; Alberti K. G. M. M.; Shaw J. Global and Societal Implications of the Diabetes Epidemic. Nature 2001, 414, 782–787. [DOI] [PubMed] [Google Scholar]
- Skyler J. S. Diabetes Mellitus: Pathogenesis and Treatment Strategies. J. Med. Chem. 2004, 47, 4113–4117. [DOI] [PubMed] [Google Scholar]
- Ahren B. Islet G Protein-coupled Receptors as Potential Targets for Treatment of Type 2 Diabetes. Nature Rev. Drug Discovery 2009, 8, 369–385. [DOI] [PubMed] [Google Scholar]
- Mohler M. L.; He Y.; Wu Z.; Hwang D. J.; Miller D. D. Recent and Emerging Anti-Diabetic targets. Med. Res. Rev. 2008, 1–71. [DOI] [PubMed] [Google Scholar]
- The details of these studies will be reported separately in the near future; also see ref (17).
- Weckbecker G.; Lewis I.; Albert R.; Schmid H. A.; Hoyer D.; Bruns C. Opportunities in Somatostatin Research: Biological, Chemical, and Therapeutic Aspects. Nature Rev. Drug Discovery 2003, 2, 999–1017. [DOI] [PubMed] [Google Scholar]
- Olias G; Viollet C.; Kusserow H.; Epelbaum J.; Meyerhof W. Regulation and Function of Somatostatin Receptors. J. Neurochem. 2004, 8951057–1091. [DOI] [PubMed] [Google Scholar]
- Florio T.; Morini M.; Villa V.; Arena S.; Corsaro A.; Thellung S.; Culler M. D.; Pfeffer V.; Noonan D. M.; Schettini G.; Albini A. Somatostatin Inhibits Tumor Angiogenesis and Growth via Somatostatin receptor-3-mediated Regulation of Endothelial Nitric Oxide Synthase and Mitogen Activated Protein Kinase Activities. Endocrinology 2003, 14441574–1584. [DOI] [PubMed] [Google Scholar]
- Srikant C. B. Cell Cycle Dependent Induction of Apoptosis by Somatostatin Analog SMS 201–995 in AtT-20 Mouse Pituitary Cells. Biochem. Biophys. Res. Commun. 1995, 2092400–406. [DOI] [PubMed] [Google Scholar]
- Lattuada D.; Casnici C.; Venuto A.; Marelli O. The Apoptotic Effect of Somatostatin Analog SMS 201-995 on Human Lymphocytes. J. Neuroimmunol. 2002, 1331–2211–216. [DOI] [PubMed] [Google Scholar]
- For a recent review of subtype selective somatostatin receptor ligands, seeWolkenberg S. E.; Thut C. J. Recent Progress in the Discovery of Selective Non-Peptide Ligands of Somatostatin Receptors. Current Opin. Drug Discovery Dev. 2008, 114446–457. [PubMed] [Google Scholar]
- Banziger M.; Cercus J.; Hirt H.; Laumen K.; Malan C.; Spindler F.; Struber F.; Troxler T. The Development of a Practical Synthesis of the Potent and Selective Somatostatin sst3 Receptor Antagonist [4-(3,4-difluoro-phenyl)-piperazine-1-yl]-{(4S,4aS,8aR)-2[(S)-3-(6-methoxy-pyridin-3-yl)-2-methyl-propyl]-decahydroisoquinoline-4-yl}-methanone (NVP-ACQ090). Tetrahedron: Asymmetry 2003, 14, 3469–3477. [Google Scholar]
- Troxler T.; Hurth K.; Schuh K.-H.; Schoeffter P.; Langenegger D.; Enz A.; Hoyer D. Decahydroisoquinoline derivatives as novel non-peptidic, potent and subtype-selective somatostatin sst3 receptor antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 1728–1734. [DOI] [PubMed] [Google Scholar]
- Poitout L.; Roubert P.; Contour-Galcera M.-O.; Moinet C.; Lannoy J.; Pommier J.; Plas P.; Bigg D.; Thurieau C. Identification of Potent Non-Peptide Somatostatin Antagonists with sst3 Selectivity. J. Med. Chem. 2001, 44, 2990–3000. [DOI] [PubMed] [Google Scholar]
- Novartis AG (Troxler T. J.; Hurth K.; Hoyer D.) β-Carboline Derivatives and its Pharmaceutical Use Against Depression and Anxiety. U.S. 6,861,430 B2, March 1, 2005.
- For details of the sst3 binding assay and the GSIS assay with pancreatic islets, see the Supporting Information and Merck & Co., Inc. (Zhou Y.-P.; Li J.; Wu W.; Shang J.; Thompson J. R.; Thornberry N. A.) Diagnosis and Treatment of Diabetes and Other Disorders. U.S. 2009/0131451A1, May 21, 2009.
- The sst3 cAMP functional assay is described in the Supporting Information.
- Wang J.; Della Penna K.; Wang H.; Karczewski J.; Connolly T. M.; Koblan K. S.; Bennett P. B.; Salata J. J. Functional and pharmacological properties of canine ERG potassium channels. Am. J. Phys. 2003, 284, H256–67. [DOI] [PubMed] [Google Scholar]
- Zhou Y.-P.; Marlen K.; Palma J. F.; Schweitzer A.; Reilly L.; Gregoire F. M.; Xu G. G.; Blume J. E.; Johnson J. D. Overexpression of Repressive cAMP Response Element Modulators in High Glucose and Fatty Acid-treated Rat Islets. J. Biol. Chem. 2003, 278, 51316–51323. [DOI] [PubMed] [Google Scholar]
- For details on the glucose tolerance test models in mice, please see the Supporting Information andTan C. P.; Feng Y.; Zhou Y.-P.; Eiermann G. J.; Petrov A.; Zhou C.; Lin S.; Salituro G.; Meinke P.; Mosley R.; Akiyama T. E.; Einstein M.; Kumar S.; Berger J. P.; Mills S. G.; Thornberry N. A.; Yang L.; Howard A. D. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes 2008, 5782211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.