

Abstract

A new series of potent TG2 inhibitors are reported that employ a 4-aminopiperidine core bearing an acrylamide warhead. We establish the structure–activity relationship of this new series and report on the transglutaminase selectivity and in vitro ADME properties of selected compounds. We demonstrate that the compounds do not conjugate glutathione in an in vitro setting and have superior plasma stability over our previous series.
Keywords: plasma stability, computer-aided drug design, SAR, acrylamides, Celiac disease, ADME
Tissue transglutaminase 2 (TG2) is a multifunctional enzyme primarily known for its calcium-dependent cross-linking activity via isopeptide bond formation.1 Less well-studied activities of TG2 include simple amidase, GTPase, ATPase, and protein disulfide isomerase activities.2−4 TG2 has been characterized in at least three forms, including open,5 closed,6 and an open-inactive form.7 Genetic deletion of TG2 in mice suggests a role for TG2 activity in mitochondrial energy function.8 TG2 overactivity has been most closely associated with Celiac disease and Huntington's disease (HD), and there is growing support for roles in inflammation and cancer.9−12
HD is an autosomal dominant, progressive neurodegenerative disease that is characterized clinically by motor, cognitive, and behavioral deficits. TG2 expression and transglutaminase activity have been shown to be increased in the brains of HD patients,13 and in vitro and in vivo studies have implicated TG2 in HD pathophysiology.14−17
Several classes of TG2 inhibitors have been reported, most of which are irreversible in nature.18 In addition, we have reported potent and selective covalent inhibitors bearing an acrylamide warhead as in 1 (Figure 1).19 Compound 1 has TG2 IC50 of 0.11 μM; however, in vitro metabolism profiling studies identified plasma stability as an issue cautioning against in vivo evaluation. Compound 1 was one of the examples showing better stability in plasma, having a half-life of 209 min in mouse plasma. Evidence supporting the acrylamide amide linkage as the site of metabolic instability was obtained by testing the plasma stability of aniline analogue 2 (Figure 1) and detecting its formation in plasma incubation of cyclopropylamide 1. Aniline 2 showed no indication of metabolism over a 24 h period in mouse plasma. The acrylamide warhead has been featured in several EGFR irreversible inhibitors that have progressed to clinical trials,20 suggesting that this moiety is not especially susceptible to metabolism and enabling compounds to reach acceptable plasma exposures in vivo. Thus, we present here a structure–activity relationship (SAR) study of a new class of acrylamide-bearing TG2 inhibitors that show improved plasma stability.
Figure 1.

Plasma stability of 1 and putative metabolite 2.
Our approach to stabilize the compounds to plasma exposure initially focused on replacing the problematic anilide with a more robust alkylamide. The compounds in Table 1 were prepared mainly to expand rapidly the SAR with respect to TG2 as this series of compounds still possessed an anilide-linked Cbz group expected to be susceptible to plasma cleavage. Upon identification of a suitable replacement, a second round of SAR would be required to replace the remaining Cbz-anilide. Our working hypothesis regarding the mode of binding of these compounds remained consistent with our prior modeling and SAR studies (Figure 2).19 Key interactions are capture of the acrylamide by Cys277 accompanied by additional acrylamide hydrogen-bonding interactions with the side chains of Trp241 and Gln276. In addition, there is a potential hydrogen-bonding interaction of the sulfonyl with the side chain of Asn333, which may facilitate further interactions with residues Leu312, Phe316, Ile331, and Leu420 that line an adjacent lipophilic pocket.
Table 1. Acrylamide Anilide Linkage Replacementsa.
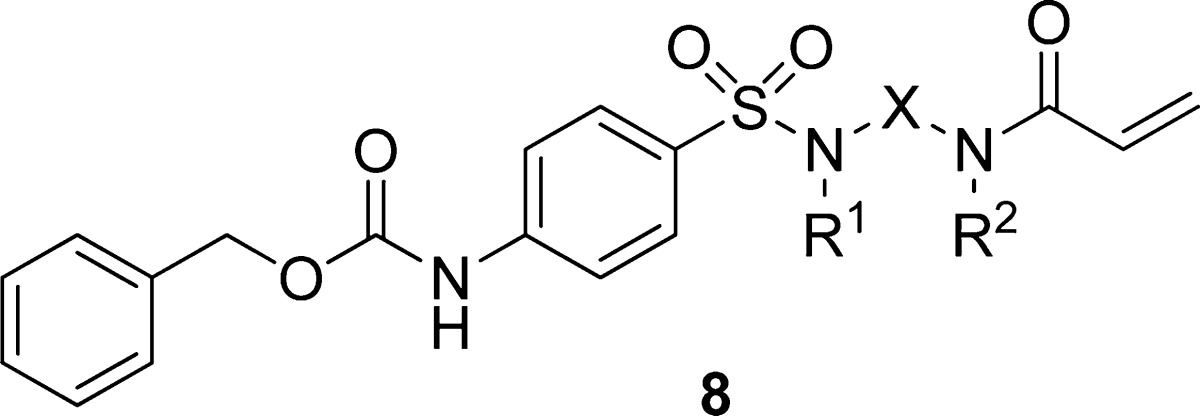
Values accompanied by standard deviations were averaged from at least two independent experiments.
Figure 2.
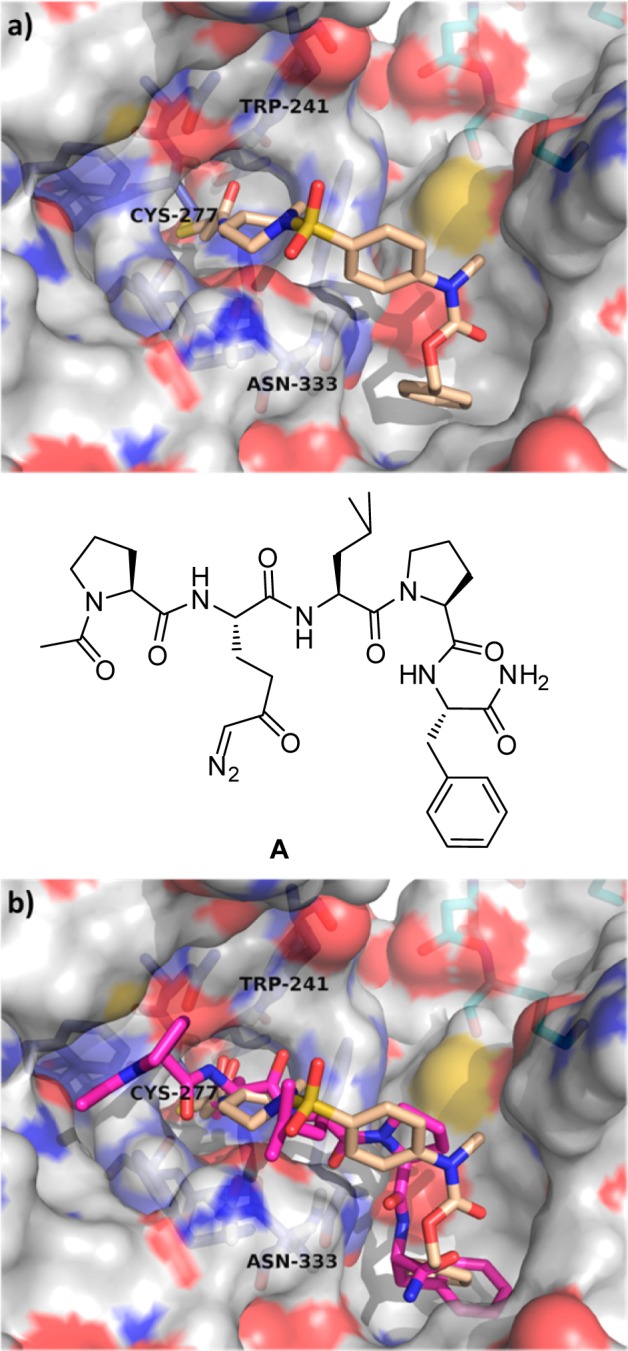
(a) Docking pose of analogue 8a (tan) covalently bound to Cys-277 within the active site of TG2. (b) Overlay of 8a with the diazoketone-containing pentapeptide inhibitor A (magenta) described by Pinkas et al.5
In our prior report,19 we established that for several human transglutaminase isoforms, a correlation exists (r2 = 0.95) between the IC50 values using a 30 min compound incubation and the irreversible inhibition constants, kinact/Ki. With this in hand, we relied on the IC50 values to guide the medicinal chemistry effort. In addition, all activity data in this report are for the human form of these enzymes unless expressly stated. As shown in Table 1, we began by preparing the 4-aminopiperidine (8a), which appeared to be of the appropriate shape and size to allow the acrylamide moiety to reach Cys277, while the sulfonamide provided a kink necessary to direct the Cbz group toward the lipophilic pocket. We were additionally intrigued that the apparent proximity of Asn333 to the sulfonamide might also contribute toward orienting the Cbz group. The TG2 activity of 8a was 2–3-fold lower with respect to anilide 1. Not surprisingly, 8a had an unacceptably short (4 h) half-life in mouse plasma. Adding a methylene as in 8b, reducing the overall distance between the sulfonamide and the acrylyl moiety as in 8c, or opening the piperazine as in 8d resulted in a 10–20-fold loss in activity. In the next set of analogues, the attachment point of the acrylamide moiety on the piperidine was changed from the 4-position to the 3-position in 8e and 8f, resulting in a complete loss of activity when tested at 80 μM. Reducing the ring size resulted in the 3-aminopyrrolidines 8g and 8h. These showed slightly improved activity, but these analogues were 100–200-fold less active when compared to 8a. Interestingly, the activity improved somewhat upon further reduction in ring size to the 3-aminoazetidine (8i). We've previously noted an inverse correlation of TG2 activity with the magnitude of acrylamide electronegativity. While we suspect that electronegativity (reactivity) is a factor contributing to TG2 potency, it is likely not an overriding feature given the 2 orders of magnitude range in potency observed within the aryl acrylamide SAR.19 Transglutaminase selectivity profiling of 8a afforded the following IC50 values: TG2, 0.28 μM; TG1, 0.91 μM; TG3, >80 μM; TG6, 8.9 μM; and FXIIIa, 3.6 μM.
The importance of the geometry and the presumed interaction of the sulfonyl moiety with Asn333 was probed using compounds 14 and 15 (Figure 3). Replacing the sulfonyl with an sp2-hybridized carbonyl resulted in 14, which had a TG2 IC50 of 8.9 μM, a 30-fold loss in activity when compared to 8a. Further reduction of the carbonyl to an sp3-hybridized methylene resulted in 15, which had a TG2 IC50 of 1.6 μM, a 6-fold loss relative to 8a. These results indicate that the effect of sp3 hybridization at this center dominates any benefit of the putative hydrogen bond with Asn333 in orienting the benzylcarbamate moiety toward the lipophilic pocket.
Figure 3.

Structures of Asn333 probes 14 and 15.
Keeping the 4-aminopiperidinyl moiety of 8a, further modifications were focused on replacement of the remaining anilide linkage. As shown in Table 2, this was accomplished by reversing the sense of the amide linkage in compounds 9a–m and ensuring in the design that only aliphatic amines were incorporated for improved stability and optimal interactions with residues Leu312, Phe316, Ile331, and Leu420 that line this portion of the binding site. Our initial compound in this study, phenethyl amide 9a, had a TG2 IC50 of 0.18 μM. Reducing the length of the ethyl linker as in 9b resulted in little change in activity; however, lengthening this linker by one methylene as in 9c resulted in a 4-fold activity loss. Reducing the number of rotatable bonds by incorporating the ethyl linker in a cyclopropane (9d) or a tetrahydoisoquinoline (9e) caused a 2–3-fold loss in TG2 activity, as did N-methylation of the amide (9f and 9g). Highlighting the lipophilic nature of this portion of the site, a small but significant improvement in activity relative to 9a was achieved by 9h, which bears a cyclohexyl moiety in place of the terminal phenyl; however, significant activity was lost upon incorporation of a larger adamantyl group (9i). This improvement was also lost in the 4-tetrahydropyranyl and morpholinyl analogues (9j and 9k), but it was nonetheless interesting to observe that the basic morpholinyl nitrogen and polar oxygen atoms were well-tolerated. The final three compounds in this set (9l–n) also attempted to reduce the number of rotatable bonds relative to 9a. These changes were mostly well-tolerated, although none possessed improved TG2 activity.
Table 2. Reversed Amide TG2 Inhibitorsa.


Values accompanied by standard deviations were averaged from at least two independent experiments.
A further expansion of the SAR around the phenethyl amide is shown in Table 3. When compared to 9a, the addition of a 4-trifluoromethyl substituent as in 9o resulted in a 3-fold loss of activity. This activity was mostly recovered in 4-chloro 9p, and a small improvement was observed in 4-methoxy 9q, which had a TG2 IC50 of 0.11 μM. The incorporation of large lipophilic groups at the 4-position, as in 9r and 9s, was disfavored, while replacement of the terminal phenyl with either a 2-naphthyl (9t) or a 7-quinolinyl moiety (9u) resulted in roughly equipotent activity. Following 9q, several alkoxy-substituted analogues (9v–y) were investigated; however, none was as potent as 9q. Of the remaining compounds, 3-chloro 9z and 3,4-dimethyl 9aa were equipotent with 9a.
Table 3. Phenethylamide Derivativesa.
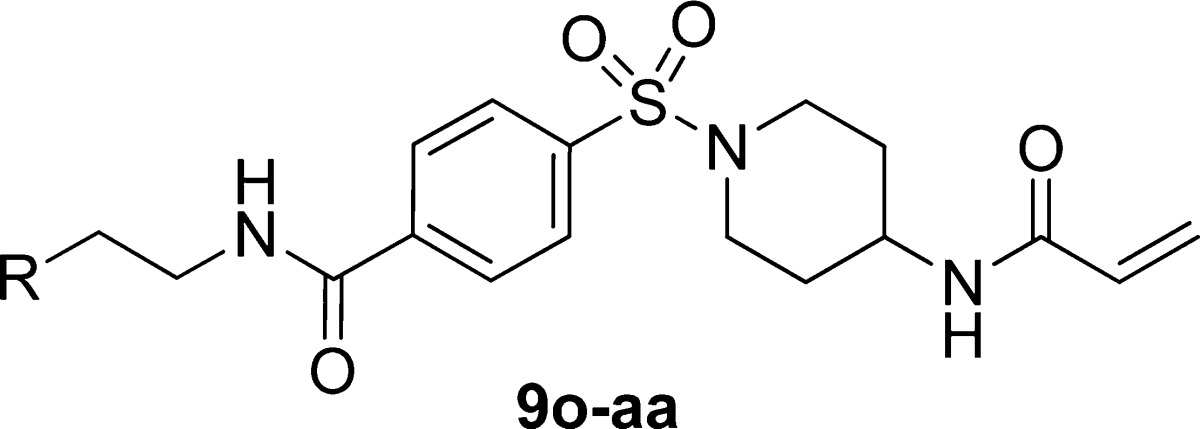
| compd | R | TG2 IC50 ± SD (μM) |
|---|---|---|
| 9a | C6H5 | 0.18 ± 0.029 |
| 9o | 4-CF3-C6H4 | 0.55 ± 0.049 |
| 9p | 4-Cl-C6H4 | 0.28 ± 0.016 |
| 9q | 4-CH3O-C6H4 | 0.11 ± 0.0028 |
| 9r | 4-t-Bu-C6H4 | 22 ± 15 |
| 9s | 4-Ph-C6H4 | 57 ± 8.8 |
| 9t | 2-naphthyl | 0.28 ± 0.0071 |
| 9u | 7-quinolinyl | 0.31 ± 0.016 |
| 9v | 2-CH3O-C6H4 | 0.23 ± 0.016 |
| 9w | 3,4-(CH3O)2-C6H3 | 0.46 ± 0.093 |
| 9x | benzo[d][1,3]dioxol-4-yl | 0.28 ± 0.052 |
| 9y | 2,3-dihydrobenzofuran-7-yl | 0.26 ± 0.081 |
| 9z | 3-Cl-C6H4 | 0.17 ± 0.021 |
| 9aa | 3,4-(CH3)2-C6H3 | 0.18 ± 0.044 |
Values accompanied by standard deviations were averaged from at least two independent experiments.
Transglutaminase selectivity profiles against mouse TG2 (mTG2) and human isoforms of TG1, TG6, and FXIIIa were obtained on the more potent compounds. The IC50 values for compound 9q were typical: TG2, 0.11 μM; mTG2, 0.055 μM; TG1, 1.2 ± 0.047 μM; TG6, 34 ± 0.062 μM; and FXIIIa, 6.4 ± 0.11 μM. It is important to note that all of the compounds tested against mTG2 had activity comparable to their human TG2 values, confirming the species-independent nature of this activity. Typical selectivity against TG1 was 3–10-fold, and >100-fold selectivity was routinely observed against TG6, while 10–20-fold selectivity was typical against FXIIIa. The TG3 activity was not tested since the compounds were expected to be inactive, this based on a large body of testing.19 The selectivity profiling revealed that further effort might be needed to improve selectivity against TG1 and FXIIIa; selectivity profiles would need to be factored into any decision to consider these compounds for in vivo studies.
Several compounds were profiled in in vitro metabolism assays of kinetic solubility, plasma stability, liver microsomal stability, and permeability/efflux. The results for compound 9a were typical; it showed aqueous solubility of <0.01 mg/mL and mouse and human liver microsomal intrinsic clearance values of 99 and 595 μL/min/mg, respectively. The high rate of metabolism was likely due to the phenethyl and aminopiperidinyl moieties, which are susceptible to oxidative metabolism at multiple sites. All compounds tested were stable in both mouse and human plasma, with a half-life >6 h and several showing a half-life >24 h. In addition, 8a and 9z showed no evidence of conjugation to the prototypical biological nucleophile glutathione (GSH) when tested in vitro over a period of 120 h.21
To be effective agents for neurodegenerative disorders, it is advantageous for compounds to possess a high rate of permeability and a low efflux rate. P-glycoprotein (P-gp) is one of the main efflux transporters in brain; the potential for compounds of interest to be effluxed by P-gp was assessed in an MDCK-MDR1 transfected cell line. Compound 9a showed a Papp A-B of 83 nm/s in MDCK-WT and an effective efflux ratio (EER) of 44, indicating moderate permeability but high active efflux via P-gp. These results were typical of all compounds tested. Combining this outcome with the results of microsomal stability testing indicated that no compound was identified as being a suitable candidate for in vivo evaluation in the context of HD. The result of this profiling suggests that the compounds may be better suited as treatments for Celiac disease, where BBB permeability would be a disadvantage and 24 h systemic coverage may not be required for efficacy.
In summary, we have developed a series of covalent TG2 inhibitors having selectivity against closely related transglutaminase isoforms exhibiting low GSH reactivity and excellent plasma stability. In vitro ADME profiling indicated rapid rates of oxidative metabolism and high P-gp efflux, suggesting that further optimization of ADME profiles will be required to achieve brain exposure in vivo.
Acknowledgments
We are indebted to Ina Sternberger for outstanding technical assistance in conducting the transglutaminase biochemical assays. We also thank Andreas Ebneth for valuable discussions.
Supporting Information Available
Synthetic disclosures including schemes and full experimental procedures and characterization of key compounds, supporting figures, details of the ADME profiling assays, and transglutaminase selectivity profiling data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Greenberg C. S.; Birckbichler R. H. Transglutaminases: Multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991, 5, 3071–3077. [DOI] [PubMed] [Google Scholar]
- Im M.-J.; Riek P.; Graham R. A novel guanine nucleotide-binding protein coupled to the α1-adrenergic receptor. J. Biol. Chem. 1990, 265, 18952–18960. [PubMed] [Google Scholar]
- Lai T.-S.; Slaughter T.; Peoples K.; Hettasch J.; Greenburg C. Regulation of human tissue transglutaminase function by magnesium-nucleotide complexes. J. Biol. Chem. 1998, 273, 1776–1781. [DOI] [PubMed] [Google Scholar]
- Hasegawa G.; Suwa M.; Ichikawa Y.; Ohtsuka T.; Kumagai S.; Kikuchi M.; Sato Y.; Saito Y. A novel function of tissue-type transglutaminase: protein disulfide isomerase. Biochem. J. 2003, 373, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkas D.; Strop P.; Brunger A.; Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLOS Biol. 2007, 5, 2788–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Cerione R. A.; Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2743–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamnaes J.; Pinkas D. M.; Fleckenstein B.; Khosla C.; Sollid L. M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroberardino P. G.; Farrace M.; Viti I.; Pavone F.; Fimia G.; Melino G.; Kodolfo C.; Piacentini M. Tissue transglutaminase contributes to the formation of disulphide bridges in proteins of mitochondrial respiratory complexes. Biochim. Biophys. Acta 2006, 1757, 1357–1365. [DOI] [PubMed] [Google Scholar]
- Molberg O.; McAdam S.; Sollid L. Role of tissue transglutaminase in celiac disease. J. Paediatr. Gastroenterol. Nutr. 2000, 30, 232–240. [DOI] [PubMed] [Google Scholar]
- Mangala L.; Mehta K. In Transglutaminases: The Family of Enzymes with Diverse Functions; Mehta K., Eckert R., Eds.; Karger: Basel, 2005; pp 125–138. [Google Scholar]
- Siegel M.; Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol. Ther. 2007, 115, 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeitner T.; Pinto J.; Krasnikov B.; Horswill M.; Cooper A. Transglutaminases and neurodegeneration. J. Neurochem. 2009, 109, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariello L.; de Cristofaro T.; Zanetti L.; Cuomo T.; Di Maio L.; Campanella G.; Rinaldi S.; Zanetti P.; Di Lauro R.; Varrone S. Transglutaminase activity is related to CAG repeat length in patients with Huntington’s disease. Hum. Genet. 1996, 98, 633–635. [DOI] [PubMed] [Google Scholar]
- Mastroberardino P. G.; Iannicola C.; Nardacci R.; Bernassola F.; DeLaurenzi V.; Melino G.; Moreno S.; Pavone F.; Oliverio S.; Fesus L.; Piacentini M. Tissue transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ. 2002, 9, 873–880. [DOI] [PubMed] [Google Scholar]
- Bailey C.; Johnson G. Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J. Neurochem. 2005, 92, 83–92. [DOI] [PubMed] [Google Scholar]
- Chun W.; Lesort M.; Tucholski J.; Faber P. W.; MacDonald M. E.; Ross C. A.; Johnson G. V. W. Tissue transglutaminase selectively modifies proteins associated with truncated mutant huntingtin in intact cells. Neurobiol. Dis. 2001, 8, 391–404. [DOI] [PubMed] [Google Scholar]
- Ruan Q.; Quintanilla R. A.; Johnson G. V. Type 2 transglutaminase differentially modulates striatal cell death in the presence of wild type or mutant huntingtin. J. Neurochem. 2007, 102, 25–36. [DOI] [PubMed] [Google Scholar]
- Keillor J. W.; Chabot N.; Roy I.; Mulani A.; Leogane O.; Pardin C.. Irreversible inhibitors of tissue transglutaminase. In Advances in Enzymology: And Related Areas of Molecular Biology; Toone E. J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2011; Vol. 78, pp 415–447. [DOI] [PubMed] [Google Scholar]
- Prime M. E.; Andersen O. A.; Barker J. J.; Brooks M. A.; Cheng R. K. Y.; Toogood-Johnson I.; Courtney S. M.; Brookfield F. A.; Yarnold C. J.; Marston R. W.; Johnson P. D.; Johnsen S. F.; Palfrey J. J.; Vaidya D.; Erfan S.; Ichihara O.; Felicetti B.; Palan S.; Pedret-Dunn A.; Schaertl S.; Sternberger I.; Ebneth A.; Scheel A.; Winkler D.; Toledo-Sherman L.; Beconi M.; Macdonald D.; Muñoz-Sanjuan I.; Dominguez C.; Wityak J. Discovery and SAR of potent and selective covalent inhibitors of transglutaminase 2 for Huntington’s disease. J. Med. Chem. 2012, 55, 1021–1046. [DOI] [PubMed] [Google Scholar]
- Carmi C.; Lodola A.; Rivara S.; Vacondio F.; Cavazzoni A.; Alfieri R. R.; Ardizzoni A.; Petronini P. G.; Mor M. Epidermal growth factor receptor irreversible inhibitors: Chemical exploration of the cysteine-trap portion. Mini Rev. Med. Chem. 2011, 11, 1019–1030. [DOI] [PubMed] [Google Scholar]
- MacFaul P. A.; Morley A. D.; Crawford J. J. A simple in vitro assay for assessing the reactivity of nitrile containing compounds. Bioorg. Med. Chem. Lett. 2009, 19, 1136–1138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.