

Abstract
Trk receptor tyrosine kinases have been implicated in cancer and pain. A crystal structure of TrkA with AZ-23 (1a) was obtained, and scaffold hopping resulted in two 5/6-bicyclic series comprising either imidazo[4,5-b]pyridines or purines. Further optimization of these two fusion series led to compounds with subnanomolar potencies against TrkA kinase in cellular assays. Antitumor effects in a TrkA-driven mouse allograft model were demonstrated with compounds 2d and 3a.
Keywords: Trk; kinase inhibitors; imidazo[4,5-b]pyridines; purines; cancer
Trk kinases belong to a subfamily of receptor tyrosine kinases, which are important components in the cell signaling cascade.1 Trks have three isoforms, TrkA, TrkB, and TrkC, and all three isoforms are activated by the growth factor ligands named neurotrophins.2 The binding of a neurotrophin to the extracellular domain of Trk triggers the phosphorylation of several key cytoplasmic tyrosine residues in the kinase domain. These tyrosine residues in turn serve as the docking sites for other downstream proteins and receptors and continue the signal transduction pathways.3
Early studies focused on the association of Trks with prostate and pancreatic cancers.4,5 However, in the past decade, increasing evidence has suggested that Trk signaling has been associated with a variety of other cancers, including neuroblastoma, breast, and colon cancers.6,7 As a consequence, the discovery of Trk kinase inhibitors has attracted considerable attention, and several classes have been disclosed.8−10 While available crystal structures for these receptors have been limited to the extracellular domains, the crystal structure of the TrkC kinase domain with a type II DFG-out oxindole inhibitor was reported this year.11
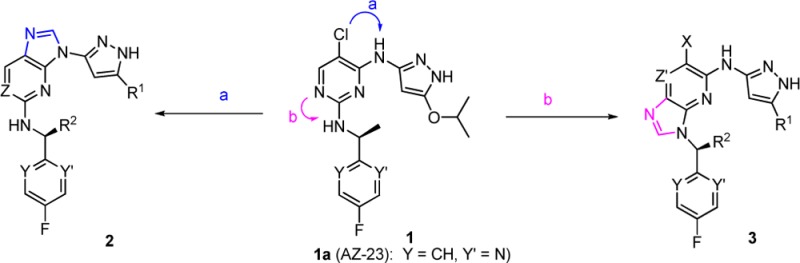
We have previously reported compound 1a (AZ-23) that showed potent TrkA/B inhibitory activities in vitro and in vivo and possessed good physical and pharmacokinetic properties.12,13 Treatment with 1a inhibited the growth of human neuroblastoma xenograft tumors, and its combination with topotecan resulted in the prolonged inhibition of tumor regrowth.14 Theoretically, compound 1a could bind to the hinge region of the kinase either through the pyrazole or through the aminopyrimidine, and modeling studies using a homology model of TrkB suggested that the pyrazole binding mode would be preferred.12 To confirm this prediction, we determined the structure of the kinase domain of TrkA in complex with 1a. TrkA adopts an inactive conformation with the αC helix in a catalytically noncompetent orientation (and the activation loop is not resolved in any of the three molecules in the asymmetric unit). Compound 1a binds through the pyrazole motif, forming hydrogen bonds to the backbone atoms of Glu 590 and Met 592 (Figure 1). The isopropoxy group is directed toward the gatekeeper Phe 589 side chain. The fluoropyridine ring contacts the side chain of Leu 657, with the fluorine atom in close proximity to the backbone carbonyl atoms of Asn 665 and the Cα of Gly 667 N-terminal to the DFG sequence of the activation loop. The pyrimidine N1 atom does not make any direct contacts with TrkA protein side chains, although it binds through water to Asp 596 NH and the flexible side chains of Asp 596 and Arg 599 in a solvent-exposed region of the ATP binding site. This heterocycle may therefore serve primarily to place the pyrazole moiety and the fluoropyridine groups appropriately.
Figure 1.
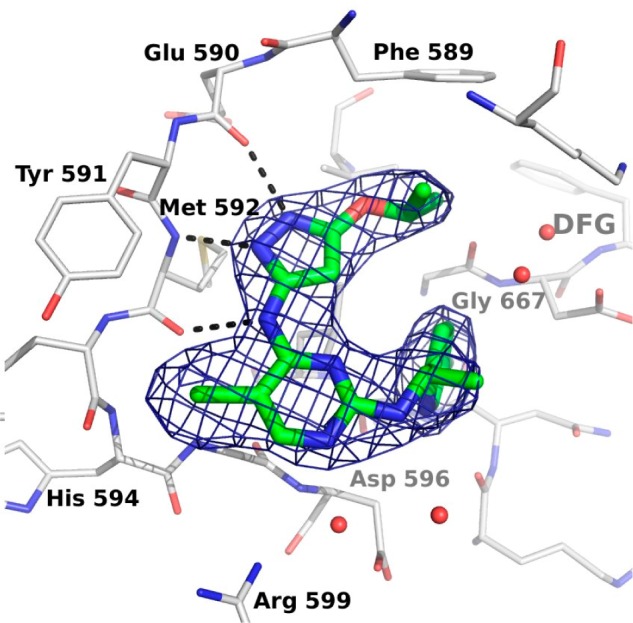
Crystal structure of TrkA with 1a. TrkA is shown in a gray stick representation, and 1a is shown in green. The weighted 2Fo – Fc map is contoured at 1.5 σ (PDB: 4AOJ).
To further probe the SAR of this ring, other scaffolds were then explored to replace the 2,4-disubstituted pyrimidine moiety. It was found that Trk inhibitory potency was retained for examples in which this ring was replaced with either phenyl, 2,6-disubstituted pyridyl, or the alternative 2,4-pyrimidine regioisomer cores.8 In addition to monocyclic central ring replacements, bicyclic ring systems were also examined. Fusions of additional rings at the C-5 and the C-6 positions on the pyrimidine ring (Figure 2) resulted in, for example, quinazoline-2,4-diamino- and thieno[2,3-e]pyrimidine-2,4-diamino- analogues that were indeed active, and their structure–activity relationship (SAR) tracked those of the monocyclic pyrimidine core.15
Figure 2.
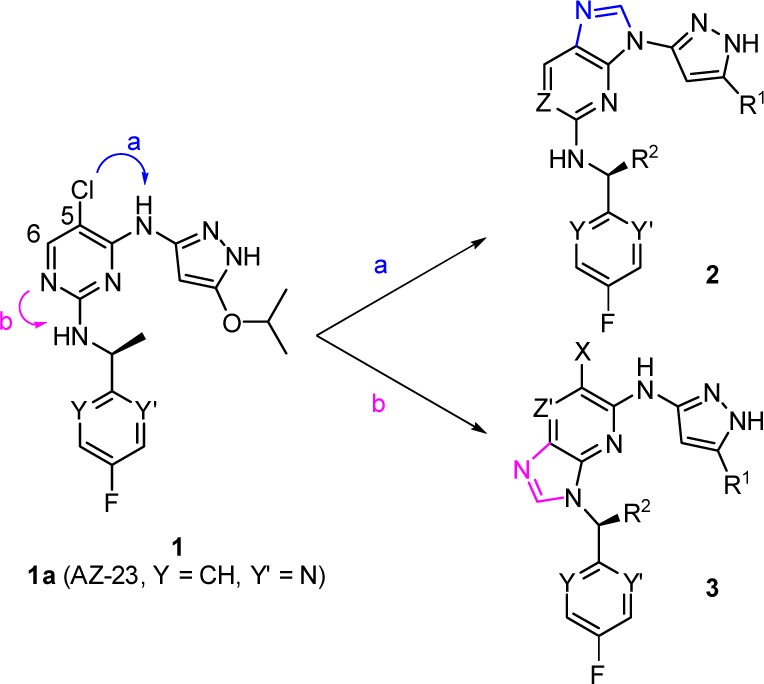
Evolution of 5/6-bicyclic scaffolds.
Two other fusion options, however, are different from the above in that one approach (Figure 2, path a) links the C-5 carbon (numbering based on 2,4-disubstituted pyrimidine system) with the exocyclic amino group of the pyrazol-3-yl to form a five-membered imidazole ring, while the second approach (Figure 2, path b) links the central ring with the nitrogen of the 1-heteroaryl ethanamino group to form another 5-membered imidazole ring. The resulting two scaffolds belong to the purine or imidazo[4,5-b]pyridine class, depending on the number of nitrogen atoms included in the 6-membered ring. It was reasoned that 3 would retain Trk potency since the pyrazol-3-yl moiety was still present after the fusion. For 2, we envisioned that the two nitrogen atoms on the pyrazole ring would still make the key hydrogen bonds as before while the third interaction might come from the weakly acidic C–H of the imidazole ring. Binding modes from docking 2d and 3a to the TrkA-1a receptor structure are illustrated in Figure 3.
Figure 3.
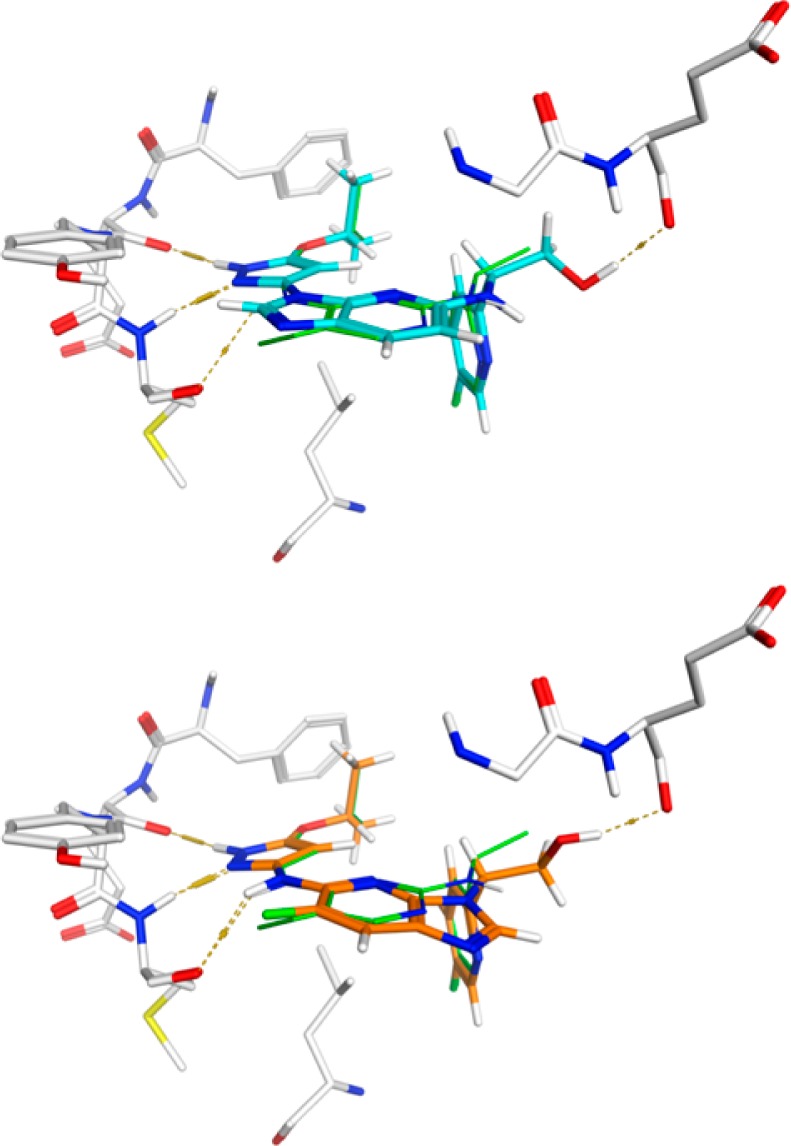
Proposed binding modes for 2d (top, cyan) and 3a (bottom, orange) in the TrkA binding site as they overlay with 1a (green). Hydrogen bonds are illustrated with dashed lines. The hydroxymethyl substituents in each example form an additional hydrogen bond with the backbone carbonyl oxygen of Glu 518 of the glycine-rich P loop.
Compounds 2a–e and 3a–c were synthesized from nitro-substituted aromatic rings, a pyrazol-3-yl amine, and a heteroaryl amine. For illustration purposes, syntheses of a pair of imidazo[4,5-b]pyridines, compounds 2d and 3a, are shown in Scheme 1. Preparation of 2d commenced from 3-nitro-2,6-dichloropyridine 4. Reaction of 4 with a pyrazol-3-yl amine 5a in the presence of triethylamine occurred regioselectively to give the intermediate 6c in 30–60% yields. A second nucleophilic aromatic substitution of intermediate 6c with an amine 7a provided 8d in 50% yield. Reduction of the nitro group of 8d, either with H2 (atmospheric balloon) in the presence of palladium on activated charcoal or alternatively with Zn/NH4Cl followed by the cyclization using formamidine acetate, resulted in the final compound 2d in 40–60% yield for two steps. Compound 2e (an aminopyrazolylpurine analogue) was synthesized in a similar fashion using 2,4-dichloro-5-nitro-pyrimidine as the starting material. Following a similar procedure to that described above for 2d, compound 3a was constructed from 9 in a few steps in good yields. It should be noted that in this case the nucleophilic aromatic substitution with the amine 7a took place prior to the pyrazole substitution with 5a.
Scheme 1. Syntheses of Compounds 2d and 3a.

Reagents and conditions: (a) CH3CN, Et3N, 30–60%. (b) BuOH, DIPEA, 40–50%. (c) Pd/C, H2, MeOH. (d) Formamidine acetate, 40–60% for two steps. (e) H2SO4, fuming HNO3, 57%. (f) EtOH, 0 °C, 75%.
Two series of compounds (2a–e and 3a–c) were profiled for in vitro cell potencies, rat in vitro/in vivo pharmacokinetics (PK), physical properties, and hERG ion channel inhibitory activities (Table 1). A constitutively active, TrkA-driven (MCF10A-TrkA-Δ) cell-based mechanism of action (MOA) assay was used as the primary cellular assay since most of these compounds had reached the detection limit for the TrkA enzyme assay (∼3 nM).12,16 The end point of this assay is the inhibition of cellular TrkA phosphorylation. Overall, these compounds exhibited subnanomolar cellular potencies. The SAR parallels that of monocyclic pyrimidine series. For example, a variety of R1 groups such as 2a (cyclopropyl), 2c (OiPr), 3b (OEt), and 3c (OCH3) were tolerated, and these analogues had subnanomolar TrkA cellular inhibitory potencies, while a smaller group such as a methyl (2b) resulted in significant loss of cellular potency. Both pyridine (2a, 2c, 2d, and 3a–c) and pyrimidine (2e) central rings offered comparable cellular potencies. At the R2 position, modeling suggested that an additional hydrogen bond could be made by polar substituents (Figure 3, 2d); however, potency was not improved as compared to 2c, likely due to competing interactions with solvent. Regarding the pharmacokinetic properties, these compounds have moderate to high clearances in rats when dosed intravenously. Intrinsic clearances (Clint) of these compounds in rat hepatocytes are also from moderate to high. Compound 2d was profiled further in beagle dogs and exhibited pharmacokinetic properties with a half-life of 1.0 h, a clearance of 15.2 mL/min/kg, and an oral bioavailability of 17%.18 The aqueous solubilities of these compounds are generally greater than 100 μM, while the unbound fraction in human plasma was measured in the range of 6–39%. The hERG ion channel inhibitory activity17 of 1a (hERG IC50: 5.2 μM) prompted us to evaluate the hERG activities of the fused ring series, and all compounds in Table 1 exhibited greater than 25 μM IC50 against this channel.
Table 1. Profiles of Compounds 2a–e and 3a–c.
| compd | R1 | R2 | Y | cella (nM) | rat hep. Clintb (μL/min/1E6) | rat iv Clc (mL/min/kg) | T1/2 (h) | F % | sola (μM) | human PPB free (%)a | hERGd (μM) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | Cp | CH3 | N | 0.24 | 48 | 39 | 1.2 | 33 | 590 | 16 | >32 |
| 2b | CH3 | CH3 | N | 281 | NT | 45 | 0.82 | 92 | >1000 | 39 | >32 |
| 2c | OiPr | CH3 | CH | 0.10 | 42 | 41 | 0.97 | 6.7 | NT | 6 | >32 |
| 2d | OiPr | CH2OH | CH | 0.50 | 13 | 46 | 2.3 | 29 | 250 | 8.1 | 52 |
| 2e | OiPr | CH3 | N | 0.24 | 24 | 130 | 3.8 | 29 | 480 | 31 | >32 |
| 3a | OiPr | 0.50 | 31 | 51 | 1.2 | 54 | 220 | 5.8 | >25 | ||
| 3b | OEt | 0.40 | 35 | 29 | 1.3 | 14 | 87 | 11 | 32 | ||
| 3c | OCH3 | 0.24 | 29 | 33 | 3.2 | 41 | 150 | 15 | >32 |
Assay conditions as in ref (12). For the cellular assay, all data are n = 1 except for 2b, which is the mean of two results.
Assay details are in the Supporting Information.
Han Wistar rats, male. Ten mg/kg po (0.1% HPMC); 3 mg/kg iv (DMA/PEG/saline = 40/40/20).
Assay conditions as in ref (17).
One question was whether compounds in series 2 might have a different selectivity profile than those in which the amino-pyrazole binding motif was intact. In an in vitro enzyme panel consisting of 82 kinases (Millipore),192c was first screened at 10 μM, and any hits (defined as kinases where >30% inhibition was seen) were followed up with dose–response curves. Compound 2c was found to inhibit 18 kinases with IC50 values less than 100 nM (Table 2). It had comparable potencies against several kinases; however, given that 2c is 10-fold more potent in the TrkA cellular assay, the limit of detection may have been reached in the Millipore assay format. The compound exhibits approximately 6-fold selectivity against FGFR1. Compound 1a had been tested in an earlier version of this assay panel (177 kinases) at 0.5 μM concentration; of kinases inhibited by >75% in this screen, dose–response curves for TrkA, TrkB, FGFR1, Flt3, Ret, MuSK, and Lck resulted in IC50 values less than 100 nM.13 While it is challenging to compare the data for 2c to the historical data for its matched pair across differing panel, concentration, and selection criteria, considering the metric of number of (non-Trk) kinases with IC50 less than 100 nM, it appears the chlorine atom at the hinge in 1a may confer additional selectivity over 2c. However, from a cellular perspective, the effect of 1a on proliferation in MCF10A-TrkA-Δ (1.51 nM) and MCF10A parental cells (2430 nM) was previously reported, with >1600-fold selectivity resulting when signaling is driven through TrkA.13 In this assay, 2c was more potent in both the TrkA-driven (0.05 nM) and the parental (192 nM) lines, with selectivity of >3800-fold.
Table 2. Kinase Selectivity Profile of 2c.
| kinase | enzyme IC50 (nM) | kinase | enzyme IC50 (nM) |
|---|---|---|---|
| Abl | 1.2 | FAK | 17.2 |
| TrkA | 1.4 | cSrc | 18.2 |
| FIt3 | 2.1 | TSSK2 | 30.6 |
| Fgr | 2.2 | ALK | 34.1 |
| BTK | 3.5 | MLK1 | 44.3 |
| SIK | 5.5 | FIt4 | 64.0 |
| PhKy2 | 6.1 | JAK2 | 68.3 |
| FGFR1 | 8.7 | Itk | 72.2 |
| SAPK2a | 13.4 | WNK3 | 99.8 |
The in vivo antitumor effects of representative examples from each series were examined in a 3T3-TrkA-Δ allograft mouse model (Supporting Information). As illustrated in Figure 4, both compounds demonstrated dose-dependent antitumor activity in this model. At 1 mg/kg dose, both 2d and 3a showed tumor growth inhibition (T/C: 12 and 30%, respectively). Compound 2d exhibited tumor stasis (T/C: −47%) at the 5 mg/kg dose, while regressions (T/C: −51 and −63%) were observed at both 5 and 25 mg/kg doses for 3a. A significant tumor growth delay (approximately 4–5 days) was seen with the 25 mg/kg dose of compound 3a. On average, minimal body weight loss (less than 5%) was seen during the dosing period in all treatment groups, including vehicle and both compounds (data not shown).
Figure 4.
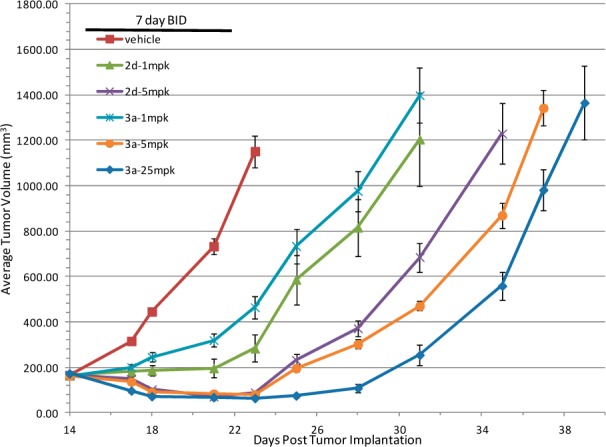
Allograft efficacies of 2d and 3a.
In summary, we have described two novel series of inhibitors against Trk kinases based on structure hopping from an earlier Trk lead. Their proposed binding modes have been illustrated through molecular docking to the first published crystal structure of the TrkA kinase domain. Each of these two fusion series could be either imidazo[4,5-b]pyridines or purines. These compounds showed potent inhibition of TrkA in a cellular assay. Assessment of the pharmacokinetic properties revealed that these compounds had moderate to high clearance in rodents and were orally bioavailable. In a TrkA-driven mouse allograft tumor model, representative compounds from both series demonstrated efficacy at doses as low as 1 mg/kg.
Acknowledgments
We thank Vladimir Capka for analytical support, Bob Young and Bin Yang for helpful discussions, and Chungang Gu for the hepatocyte assay. Kicki Blahò and Fredrik Lindqvist purified TrkA for crystallization, and Margareta Ek is acknowledged for initial crystallization screening.
Supporting Information Available
Experimental procedures regarding TrkA protein purification and crystallography, molecular docking, the rat hepatocyte clearance assay, efficacy studies, synthetic procedures, and analytical data for 2a–e and 3a–c. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Centaurus BioPharma Co., Ltd., Building #16, Yuquan Wisdom Vale, Minzhuang Road #3, Beijing 100195, China.
Author Present Address
⊥ Sirtris, A GSK Company, 200 Technology Square, Cambridge, Massachusetts 02139, United States.
Author Present Address
# Peloton Therapeutics, Inc., 2330 Inwood Road, Suite 226, Dallas, Texas 75235, United States.
Author Present Address
∇ Array BioPharma Inc., 3200 Walnut Street, Boulder, Colorado 80301, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. The authors declare no competing financial interest.
The authors declare no competing financial interest.
Supplementary Material
References
- Gschwind A.; Fischer O. M.; Ullrich A. The Discovery of Receptor Tyrosine Kinases: Targets for Cancer Therapy. Nat. Rev. Cancer 2004, 4, 361–370. [DOI] [PubMed] [Google Scholar]
- Patapoutian A.; Reichardt L. F. Trk Receptors: Mediators of Neurotrophin Action. Curr. Opin. Neurobiol. 2001, 11, 272–280. [DOI] [PubMed] [Google Scholar]
- Pattarawarapan M.; Burgess K. Molecular Basis of Neurotrophin-Receptor Interactions. J. Med. Chem. 2003, 46, 5277–5291. [DOI] [PubMed] [Google Scholar]
- Sclabas G. M.; Fujioka S.; Schmidt C.; Li Z.; Frederick W. A. I.; Yang W.; Yokoi K.; Evans D. B.; Abbruzzese J. L.; Hess K. R.; Zhang W.; Fidler I. J.; Chiao P. J. Overexpression of Tropomysin-Related Kinase B in Metastatic Human Pancreatic Cancer Cells. Clin. Cancer Res. 2005, 11, 440–449. [PubMed] [Google Scholar]
- Weeraratna A. T.; Arnold J. T.; George D. J.; DeMarzo A.; Isaacs J. T. Rational Basis for Trk Inhibition Therapy for Prostate Cancer. Prostate 2000, 45, 40–48. [DOI] [PubMed] [Google Scholar]
- Thiele C. J.; Li Z.; McKee A. E. On Trk—The TrkB Signal Tranduction Pathway Is an Increasingly Important Target in Cancer Biology. Clin. Cancer Res. 2009, 15, 5962–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur G. M.; Minturn J. E.; Ho R.; Simpson A. M.; Iyer R.; Varela C. R.; Light J. E.; Kolla V.; Evans A. E. Trk Receptor Expression and Inhibition in Neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, seeWang T.; Yu D.; Lamb M. L. Trk Kinase Inhibitors as New Treatments for Cancer and Pain. Expert Opin. Ther. Pat. 2009, 19, 1–15. [DOI] [PubMed] [Google Scholar]
- Albanese C.; Alzani R.; Amboldi N.; Avanzi N.; Ballinari D.; Brasca M. G.; Festuccia C.; Fiorentini F.; Locatelli G.; Pastori W.; Patton V.; Roletto F.; Colotta F.; Galvani A.; Isacchi A.; Moll J.; Pesenti E.; Mercurio C.; Ciomei M. Dual Targeting of CDK and Tropomyosin Receptor Kinase Families by the Oral Inhibitor PHA-848125, an Agent with Broad-Spectrum Antitumor Efficacy. Mol. Cancer Ther. 2010, 9, 2243–2254. [DOI] [PubMed] [Google Scholar]
- Ghilardi J. R.; Freeman K. T.; Jimenez-Andrade J. M.; Mantyh W. G.; Bloom A. P.; Kuskowski M. A.; Mantyh P. W. Administration of a Tropomyosin Receptor Kinase Inhibitor Attenuates Sarcoma-induced Nerve Sprouting, Neuroma Formation and Bone Cancer Pain. Mol. Pain 2010, 6, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaugh P.; Fan Y.; Mi Y.; Sun F.; Adrian F.; Li N.; Jia Y.; Sarkisova Y.; Kreusch A.; Hood T.; Lu M.; Liu G.; Huang S.; Liu Z.; Loren J; Tuntland T.; Karanewsky D. S.; Seidel H. M.; Molteni V. Discovery of GNF-5837, a Selective TRK Inhibitor with Efficacy in Rodent Cancer Tumor Models. ACS Med. Chem. Lett. 2012, 3, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Lamb M. L.; Scott D. A.; Wang H.; Block M. H.; Lyne P. D.; Lee J. W.; Davies A. M.; Zhang H.-J..; Zhu Y.; Gu F.; Han Y.; Wang B.; Mohr P. J.; Kaus R. J.; Josey J. A.; Hoffmann E.; Thress K.; MacIntyre T.; Wang H.; Omer C. A.; Yu D. Identification of 4-Aminopyrazolylpyrimidines as Potent Inhibitors of Trk Kinases. J. Med. Chem. 2008, 51, 4672–4684. [DOI] [PubMed] [Google Scholar]
- Thress K.; MacIntyre T.; Wang H.; Whitston D.; Liu Z.-Y.; Hoffmann E.; Wang T.; Brown J. L.; Webster K.; Omer C.; Zage P. E.; Zeng L.; Zweidler-McKay P. A. Identification and Preclinical Characterization of AZ-23, a Novel, Selective, and Orally Bioavailable Inhibitor of the Trk Kinase Pathway. Mol. Cancer Ther. 2009, 8, 1818–1827. [DOI] [PubMed] [Google Scholar]
- Zage P. E.; Graham T. C.; Zeng L.; Fang W.; Pien C.; Thress K.; Omer C.; Brown J. L.; Zweidler-McKay P. A. The Selective Trk Inhibitor AZ623 Inhibits Brain-Derived Neurotrophic Factor-Mediated Neuroblastoma Cell Proliferation and Signaling and is Synergistic with Topotecan. Cancer 2011, 117, 1321–1329. [DOI] [PubMed] [Google Scholar]
- Block M. H.; Han Y.; Josey J. A.; Lee J. W.; Scott D.; Wang B.; Wang H.; Wang T.; Yu D.. WO200549033, 2005.
- Angeles T. S.; Lippy J. S.; Yang S. X. Quantitative, High-Throughput Cell-Based Assays for Inhibitors of TrkA Receptor. Anal. Biochem. 2000, 278, 93–98. [DOI] [PubMed] [Google Scholar]
- Bridgland-Taylor M. H.; Hargreaves A. C.; Easter A.; Orme A.; Henthorn D. C.; Ding M.; Davis A. M.; Small B. G.; Heapy C. G.; Abi-Gerges N.; Persson F.; Jacobson I.; Sullivan M.; Albertson N.; Hammond T. G.; Sullivan E.; Valentin J. P.; Pollard C. E. Optimisation and validation of a medium-throughput electrophysiology-based hERG assay using IonWorks HT. J. Pharmacol. Toxicol. Methods 2006, 54, 189–199. [DOI] [PubMed] [Google Scholar]
- Beagle dogs, male. Three mg/kg po (25% HP-β-cyclodextrin); 2 mg/kg iv (25% HP-β-cyclodextrin).
- http://www.millipore.com/drugdiscovery/dd3/KinaseProfiler.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.