

Abstract
Triage of a set of antimalaria hit compounds, identified through high throughput screening against the Chloroquine sensitive (3D7) and resistant (Dd2) parasite Plasmodium falciparum strains identified several novel chemotypes suitable for hit-to-lead chemistry investigation. The set was further refined through investigation of their in vitro ADME properties, which identified templates with good potential to be developed further as antimalarial agents. One example was profiled in an in vivo murine Plasmodium berghei model of malaria infection.
Keywords: Screening, Plasmodium falciparum, phenotypic screening, hit-to-lead chemistry
About 3.3 billion people, half of the world’s population, are at risk of malaria. This leads to about 250 million malaria cases and nearly one million deaths annually.1 People living in the poorest countries are the most vulnerable. Malaria is an especially serious problem in Africa, where one in every five childhood deaths is due to the effects of the disease. An African child has on average between 1.6 and 5.4 episodes of malaria fever each year. Worldwide, a child dies from malaria every 30 s.1 The disease is caused by parasites of the genus Plasmodium. Plasmodium falciparum and Plasmodium vivax are predominantly responsible for the highest mortality and highest morbidity, respectively.2 Current therapy, while effective, is suffering from increased incidence of drug resistance; plus, there have been no new chemical classes of antimalarials introduced into clinical practice since 1996.3 Taken together, the discoveries of new antimalarial agents with novel modes of action are desirable.
During the last five years there has been a significant increase in activity in the antimalarial discovery arena with Novartis,4 St Judes Children’s Research Hospital Memphis,5 and GlaxoSmithKline6 making major contributions to the body of publicly available compound data.
The Medical Research Council Technology’s compound library of 48,000 compounds was screened using a phenotypic image based antimalarial high-throughput screening (HTS) assay developed and performed within the Discovery Biology group at Griffith University.7 From the primary screening, 389 compounds were identified with >40% inhibition against the Chloroquine sensitive 3D7 strain. These hits were subsequently retested against the 3D7 and resistant (Dd2) Plasmodium falciparum strains, as well as a HEK-293 cell line for mammalian cytotoxicity. After retesting, thirty compounds were found to be equally active against both parasite strains, having IC50 values <1 μM and no observable cytotoxicity at 4 μM.
The thirty most active compounds from the screen were subdivided into twenty structural classes, which were then reviewed and refined on the basis of potential for the generation of structural analogues in addition to feedback from MMV indicating which series were already being explored by other organizations or had historically been unsuccessful candidates for antimalaria drug discovery efforts.
As a result, the list was prioritized to thirteen active compounds in eight distinct classes (see Figure 1), quinazolines 1, pyrazolopyridazines 2, benzoxathiazoles 3, sulfonamides 4, thienopyrimidines 5, sulfamides 6, acyl guanidines 7, and benzimidazoles 8.
Figure 1.

Eight distinct classes of active anti-malarials after HTS and review.
Eleven of the thirteen active compounds were available for repurchase as solid samples, and an additional forty-six structural analogues (selected to establish rudimentary SAR within each series) were also sourced from commercial suppliers (see the Supporting Information for full details and QC). The purity and structures of all 57 compounds were confirmed and then assayed at the Swiss Tropical and Public Health Institute against the Plasmodium falciparum strain NF54 (3D7 is a clone of NF54 and is Chloroquine sensitive) using incorporation of 3H-Hypoxanthine as the primary readout.
Of the eight classes of compounds rescreened, four classes (benzoxathiazoles 3, sulfonamides 4, quinazolines 1, and sulfamides 6) were abandoned due to either weak or lack of inhibitory activity of the original hits or close analogues or poor whole cell ligand efficiency.8
From the four classes selected for progression, the pyrazolopyridazines (Table 1) demonstrate reasonable SAR. SAR data clearly indicates that the presence of the chloro substituent in the 4 position is essential for activity and replacement with isosteric methyl, ethoxy, or morpholine substituents, resulting in loss of all activity. The presence of the seemingly essential chlorine atom adjacent to the pyridazine nitrogen represented a potential downstream development risk, and as such, this series of molecules was down-prioritized.
Table 1. SAR Pyrazolopyridazines 9–18.


Values are a mean of ≥2 experiments in all tables unless otherwise stated.
Reference (8).
The guanadinyl substituted quinazoline was investigated (Table 2), and although the potency of this series of molecules was modest, the whole cell ligand efficiency values indicated that they may be reasonable starting points for a hit-to-lead effort, with all examples displaying ligand efficiency indices over 0.3. However, this series was down prioritized due to the presence of the guanidine functionality and the associated likely permeability risk (even though heteroaryl guanidine has lower pKa) and relatively weak potency.9
Table 2. SAR of Acyl Guanidines 19–22.


Values are a mean of ≥2 experiments in all tables unless otherwise stated.
Reference (8).
The benzimidazole containing hit molecules represents a good starting point for hit-to-lead chemistry due to the expected relative ease of synthesis of the hits and close analogues. The series shows good activity in the original HTS (1–100 μM) and clear SAR with simple substituted amines in the 4-position preferred (Table 3). Acylation and sulfonylation (compounds 23, 24, and 28) of the 4-amino substituent results in loss of activity. However, molecules with simple substituted benzyl groups have respectable whole cell ligand efficiencies (LE). A search of the literature and a robust discussion with MMV indicated that there was adequate freedom to operate for further exploration.
Table 3. SAR of Benzimidazoles 23–28.


Values are a mean of ≥2 experiments in all tables unless otherwise stated.
Reference (8).
The thienopyrimidine subclass of molecules was examined. This series demonstrates clear SAR with a relatively wide level of diversity tolerated in the heterocyclic substituent in the 4-position of the thienopyrmidine ring including monocyclic and fused bicyclic derivatives (see Table 4).
Table 4. SAR of Thienopyrimidines 29–35.


Values are a mean of ≥2 experiments in all tables unless otherwise stated.
Reference (8).
However, there appears to be a decrease in potency with substitution on the 2- and 3-positions of the thienopyrimidine ring 34 and 35. In all cases, the LE supports the fact that this series of molecules represent a good starting point for further optimization. Literature searches indicated excellent freedom to operate for this series which was confirmed by MMV.
On the basis of the literature searches, potency, demonstration of SAR, and whole cell ligand efficiency, the benzimidazoles and thienopyrimidines were selected for further investigation and an example from each series was chosen for determination of some critical in vitro ADME parameters (Table 5).
Table 5. Selected ADME Data.
| 33 | 27 | |
|---|---|---|
| solubility (μM)a | 20 | <5 |
| log D (pH 7.4)b | >3.8 | 2.0 |
| human microsomal stab. (% turnover, 40 min)c | 50 | 19 |
| permeability (A > B [10–6 cm/s])d | 50 | 1.5 |
| efflux ratio | 0.28 | 0.43 |
Kinetic solubility in phosphate buffered solution (PBS) at pH 7.4.
Partition coefficient determined between ocatan-1-ol and phosphate buffered PBS at pH 7.4.
1 μM test concentration was carried out with pooled microsomes (0.25 mg protein/mL).
Apical and basal chambers were prepared in Hanks Balanced Salt Solution (HBSS) at pH 7.4. Compounds (10 μM) were added to the donor at time zero. Both the donor and acceptor chamber were sampled following 60 min of incubation at 37 °C.
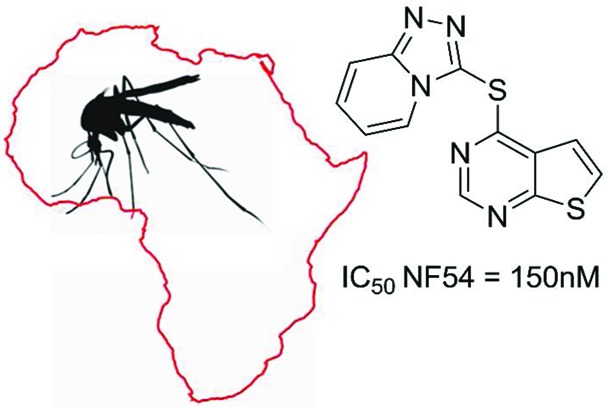
As expected, the triazole-thienopyrimidine 33 had moderate solubility (20 μM) and the benzimidazole 27 showed poor solubility (<10 μM; which may be addressed by decreasing the lipophilicity of the compounds). The triazole-thienopyrimidine had moderate microsomal turnover (50% remaining after 40 min exposure), possibly due to the sulfur linker, but the benzimidazole was metabolically stable, with a metabolic turnover in human liver microsomes of only 19% after 40 min. The triazole-thienopyrimidine showed good permeability while the benzimidazole showed poor permeability and low mass balance, which may be reflective of the low compound solubility.
The most attractive compound from this initial study in terms of antiplasmodial activity and in vitro ADME properties, 33, was assessed in vivo in the Plasmodium berghei infected mouse model. The ability of the compound to reduce the parasitemia was evaluated by dosing compound 33 at 50 mg/kg subcutaneously (sc) once a day for 4 days (Peters test). This dosing regimen resulted in a 34% reduction in parasitemia over the course of the experiment. Also, no gain in survival days compared to the case of infected, untreated control animals was observed. This level of activity is marginal in terms of statistical significance but is of sufficient interest to warrant further investigation into this series. Specifically, given that the in vitro ADME properties of both compounds studied are moderate, there is sufficient justification to suppose that optimization of these parameters will lead to an improved in vivo DMPK profile and hence improved efficacy of these compound classes when assessed in vivo. Also, both series demonstrate SAR, which appears not to be linked to lipophilicity, giving further confidence that solubility and microsomal stability can be optimized. Future optimization plans will focus on improving the in vitro ADME profile compounds focusing on reducing the cLogP and improving the solubility of the series.
In summary, a HTS campaign identified 389 novel antiplasmodial compounds with 30 examples demonstrating sub-micromolar IC50’s against resistant and nonresistant strains of Plasmodium falciparum with good selectivity indices against cytotoxicity. The genesis of the hit identification; hit confirmation; resistance and cytotoxicity profiling; analogue sourcing; SAR analysis; ADME studies; and finally in vivo assessment is summarized diagrammatically in Figure 2. Preliminary SAR refined the hits to two promising series of compounds with good freedom to operate and the potential for further follow-up through optimization of their ADME properties.
Figure 2.

Schematic representation of compound progression to in vivo assessment.
Acknowledgments
The authors thank M.M.V. for advice on compound selection.
Supporting Information Available
Spectral data and vendors for compounds 9–35 and details regarding biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Greenwood B. M.; Fidock D. A.; Kyle D. E.; Kappe S. H. I.; Alonso P. L.; Collins F. H.; Duffy P. E. Malaria: Progress, Perils and Prospects for Eradication. J. Clin. Invest. 2008, 118, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organisation. World malaria report; http://www.who.int/malaria/world_malaria_report_2011/en/.
- Wells T. N.; Poll Elizabeth M. When is enough enough? The need for a robust pipeline of high-quality antimalarials. Discovery Med. 2010, 948389–98. [PubMed] [Google Scholar]
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrian F.; Matzen J. T.; Anderson P.; Nam T. G.; Gray N. S.; Chatterjee A. K.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. 2008, 105, 9059–9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jimenenz-Diaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; El Mazouni F.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; De Risi J. L.; Sullivan D. J. Jr.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W.; Avery V. M.; Guy R. K. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F.-J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J.-L.; Vanderwall D. E.; Green D. V. S.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [DOI] [PubMed] [Google Scholar]
- Duffy S.; Avery V. M. Development and Optimization of a Novel 384-Well Anti-Malarial Imaging Assay Validated for High-Throughput Screening. Am. J. Trop. Med. Hyg. 2012, 86184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alex A.; Hopkins A. L.; Groom C. R. Ligand efficiency: A useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Humphreys W. G.; Obermeier M. T.; Chong S.; Kimball S. D.; Das J.; Chen P.; Moquin R.; Han W.-C.; Gedamke R.; White R. E.; Morrison R. A. Oxidative activation of acylguanidine prodrugs: intestinal presystemic activation in rats limits absorption and can be inhibited by co-administration of ketoconazole. Xenobiotica 2003, 33193–106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.