

Abstract

A new series of cyclic sulfamide derivatives were synthesized and evaluated for their ability to inhibit 11β-HSD1. Among this series, 18e showed good in vitro activity toward human 11β-HSD1, selectivity against 11β-HSD2, microsomal stability, and pharmacokinetic and safety profiles (hERG, CYP, and acute toxicity). Additionally, 18e exhibited good in vivo efficacy in rat and monkey models.
Keywords: diabetes, antidiabetic agents, 11β-hydroxysteroid dehydrogenase type 1, cyclic sulfamide, adamantyl group
An endoplasmic reticulum-associated enzyme, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), acts predominantly as an NADPH-dependent reductase in vivo and converts inactive cortisone to the active glucocorticoid cortisol1 (Figure 1).
Figure 1.

Role of 11β-HSD1 between cortisone and cortisol.
The relationship between 11β-HSD1 and type 2 diabetes has been demonstrated in genetic mouse models. Mice overexpressing 11β-HSD1 in adipose tissues showed metabolic syndrome-like phenotypes such as central obesity, glucose intolerance, and insulin resistance.2,3 In contrast, 11β-HSD1-deficient mice were resistant to the development of high-fat diet-induced obesity and exhibited improved insulin sensitivity and lipid profiles.4,5 These data suggest that 11β-HSD1 could be a drug target for the treatment of metabolic syndrome as well as type 2 diabetes. During the past few years, a number of small molecule 11β-HSD1 inhibitors have been reported,6−15 and several candidates including Incyte's compound are in clinical trials.8
Among the classes of 11β-HSD1
inhibitors, the adamantyl group is one of the most popular and promising
skeletons.9−13 Therefore, we looked for a new 11β-HSD1 inhibitor with an
adamantyl group and found a promising cyclic sulfamide skeleton with
an adamantyl group.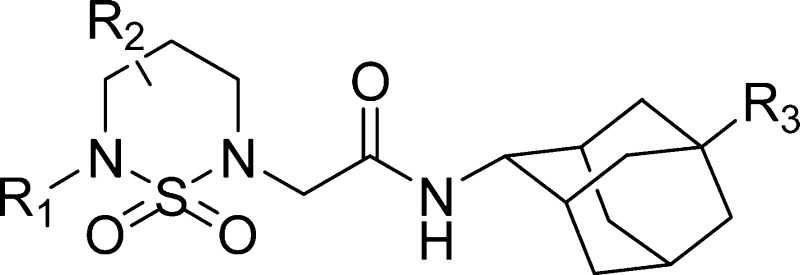
We now report the synthesis of cyclic sulfamide derivatives with an adamantyl group and their biological evaluation as 11β-HSD1 inhibitors.
A series of cyclic sulfamide derivatives were synthesized according to Schemes 1–3. Chlorosulfonyl isocyanate 1 was reacted sequentially with tert-butyl alcohol and chloropropylamine to provide 2, which was cyclized under basic condition to give Boc-cyclic sulfamide 3. Compound 3 was alkylated with ethyl bromoacetate to produce compound 4. Alkaline hydrolysis then provided an acid, which was amidated with 2-adamantyl amine to obtain compound 5. Compound 5 was deprotected by 4 M HCl and alkylated to produce the arylalkyl cyclic sulfamide derivatives 6.
Scheme 1.

Scheme 3.

N-Phenyl cyclic sulfamide derivatives were synthesized according to Scheme 2. Sulfuryldichloride 7 was coupled with 3-chloropropylamine and anilines to give compound 9, which was cyclized under basic condition to give 10. Compound 10 was coupled with ethyl bromoacetate to give 11, which was hydrolyzed and amidated by adamantyl amines to produce final compound 13.
Scheme 2.

Analogues bearing substitution on the sulfamide-containing ring were obtained as shown in Scheme 3. Sulfuryl dichloride 7 was coupled with glycine ethyl ester and anilines to give compound 15, which was N-substituted by alkylation or Mitsunobu reaction to obtain compound 16. Compound 16 was then hydrolyzed and coupled with adamantyl amines to produce the final compound 18.
In vitro inhibition activity of 11β-HSD1 was assessed by a HTRF cortisol concentration assay. Human and mouse 11β-HSD1 overexpressed cells were incubated with cortisone and each compound for 3 h. The IC50 values of the compounds were determined from concentration-dependent inhibition curves. Carbenoxolone was used as a standard 11β-HSD1 inhibitor.
First, Boc-protected cyclic sulfamide derivative with 2-adamantyl group (5) showed nanomolar inhibitory activity with an IC50 value of 363 nM toward 11β-HSD1. Therefore, we further modified sulfamide scaffold by replacing the Boc group. Instead of Boc, the benzyl substituent 6a showed similar activity as that with Boc; however, the phenethyl substituent 6b displayed weak inhibitory activity (1.66 μM). Indeed, phenyl substituent 13a showed good in vitro activity with an IC50 value of 12 nM. However, 1-adamantyl derivative 13b and acid compound 12 were weakly active and not active, respectively.
Table 1. In Vitro Human 11β-HSD1 Inhibitory Activity of Cyclic Sulfonamide Derivatives.
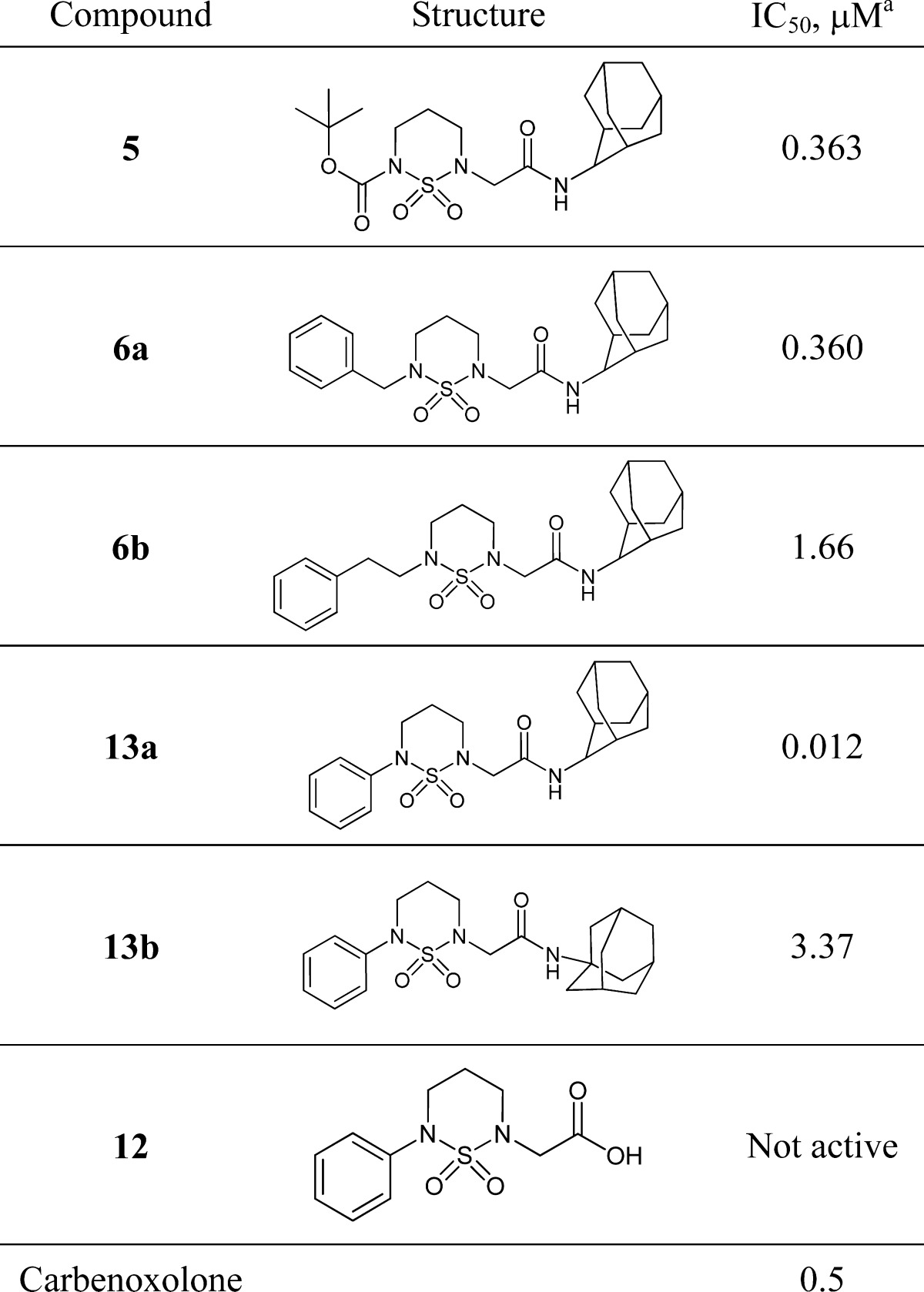
IC50 values were determined by GraphPad Prism software. Results are expressed as means ± SEMs of triplicate experiments.
We focused our attention on N-phenylsubstituted cyclic sulfamide with adamantylcarboxamide (13c), which has better microsomal stability than unsubstituted adamantyl group,16 and the results are summarized in Table 2. Compounds 13c–h are E isomers, which showed better in vitro activity than the Z isomers; therefore, we focused on the E isomer. Unsubstituted phenyl derivative with adamantylcarboxamide (13c) showed good in vitro activity with an IC50 value of 17 nM for human 11β-HSD1 and moderate activity with an IC50 value of 207 nM for mouse 11β-HSD1. Although 3-fluoro (13e), 4-fluoro (13f), and 2,4,6-trifluorophenyl (13g) derivatives showed moderate to good in vitro activities, 2,4,6-trichlorophenyl derivative (13h) was the most active in this series with 1 and 4 nM for human and mouse 11β-HSD1, respectively.
Table 2. In Vitro 11β-HSD1 Inhibitory Activity of Cyclic Sulfonamide Derivatives.
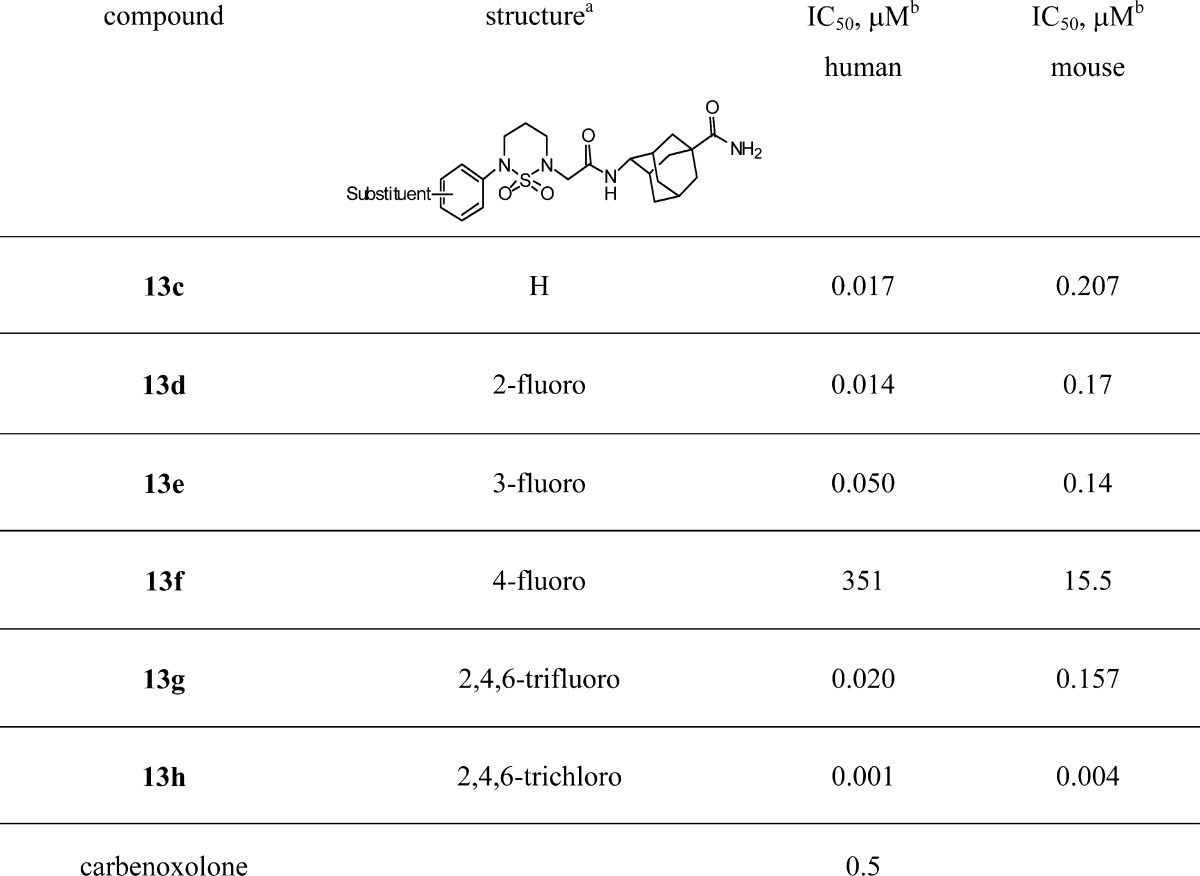
(E)-Adamantylcarboxamide isomer.
IC50 values were determined by GraphPad Prism software.
The compound 13h showed good in vitro activities; therefore, we performed in vivo 11β-HSD1 inhibition study in normal mice. After 20 mpk oral dosing, 11β-HSD1 inhibition was measured in fat and liver tissues. Compound 13h showed 86 and 85% 11β-HSD1 inhibitions in the fat and liver tissues after 2 h, respectively. However, human and rat liver microsomal stabilities of 13h were moderate with 61 and 57% of the parent compound remaining after 30 min of incubation. To improve the liver microsomal stability, we changed the six-membered ring to five- or seven-membered ring and ring-opening structure. Unfortunately, the five-memered ring (18a) exhibited reduced activity with submicromolar potency. Moreover, although the seven-membered ring (18b) and ring-opened dimethyl derivative (18c) showed good in vitro potencies, their in vivo 11β-HSD1 inhibitions and liver microsomal stabilities were not improved.
Table 3. In Vitro 11β-HSD1 Inhibitory Activity, Liver Microsomal Stability, and 11β-HSD Inhibition of Cyclic Sulfonamide Derivatives.
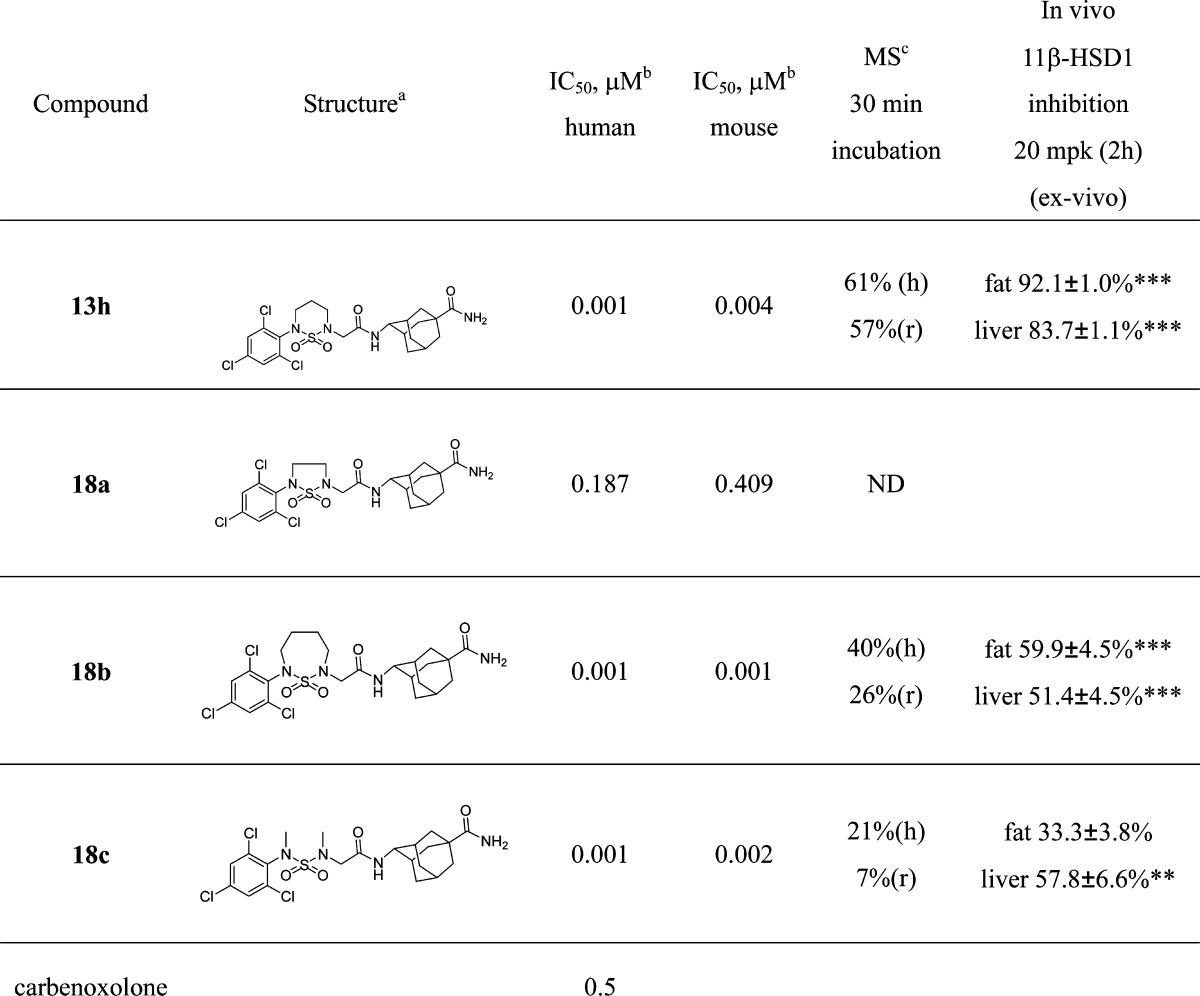
(E)-Adamantylcarboxamide isomer.
IC50 values were determined by GraphPad Prism software.
Liver microsomal stability. Results of ex vivo 11β-HSD1 inhibition are expressed as means ± SEMs for n = 4 mice per group. **P < 0.01, ***P < 0.001 vs vehicle group.
Therefore, we further modified the six-membered ring with diverse substituents, and the results are summarized in Table 4. 2-Methyl substituent (18d) showed improved metabolic stability in human and rat liver microsome. Furthermore, 18e was the most potent in this series exhibiting good in vitro activities with 1 and 2 nM toward human and mouse 11β-HSD1, respectively, as well as liver microsomal stability (93 and 78% after 30 min of incubation). Compound 18e exhibited the best in vivo 11β-HSD1 inhibition efficacy with 95% inhibition after 20 mpk oral administration.
Table 4. In Vitro 11β-HSD1 Inhibitory Activity, Liver Microsomal Stability, and 11β-HSD 1 Inhibition of Cyclic Sulfonamide Derivatives.

(E)-Adamantylcarboxamide isomer.
IC50 values were determined by GraphPad Prism software.
The chirality of methyl is racemic. Results of in vivo 11β-HSD1inhibition are expressed as means ± SEMs for n = 4 mice per group. **P < 0.01, ***P < 0.001 vs vehicle group.
As shown in Table 5, 18e exhibited good selectivity against 11β-HSD2 and plasma stability. Additionally, 18e showed no significant inhibition any of the major CYP isoforms (main cytochrome P450 enzymes, 1A2, 2C9, 2C19, 2D6, and 3A4). Moreover, it showed weak inhibition of the hERG channel (36.9 μM), reasonable solubility (361 μM), and a LD50 value of over 1000 mpk.
Table 5. Selectivity, Stability, CYP Inhibition, hERG, Solubility, and Acute Toxicity of 18e.
| entry | hHSD2 inhibition | plasma stability | CYP inhibition | hERG | aqueous solubility | acute toxicity |
|---|---|---|---|---|---|---|
| 18e | 20% at 10 μM | 100% after 30 min incubation | 1A2 > 100 μM | 36.9 μM | 361 μM | LD50 > 1000 mpk |
| 2C9 > 100 μM | ||||||
| 2C19 > 100 μM | ||||||
| 2D6 > 100 μM | ||||||
| 3A4 > 100 μM |
The PK and PD profiles of 18e were evaluated in rat and monkey and are summarized in Table 6. Compound 18e showed good bioavailability (69%) and moderate clearance (CL = 0.7) in rat. The compound 18e significantly and dose dependently reduced 11β-HSD1 activity in fat tissues in 2 h by 77, 90, 91, and 96% at 1, 5, 10, and 20 mpk, respectively. Additionally, a nonhuman primate (cynomologous monkey) was dosed orally with 18e at 10 mpk and exhibited over 80% 11β-HSD1 inhibition in three fat depots in 2 h and a good blood exposure level.
Table 6. In Vivo PK and PD Study of 18ea.
| rat PK (10 mpk) | in vivo 11β-HSD1 inhibition (po) in mice (ex vivo) | monkey PK (10 mpk) | in vivo 11β-HSD1 inhibition (po) in monkey (ex vivo) |
|---|---|---|---|
| po Cmax = 3.1 μg/mL | (1 mpk) fat (76.6 ± 2.8%) | po Cmax = 6.4 ug/mL | 10 mpk |
| t1/2 = 3.8 h | (5 mpk) fat (90.4 ± 0.6%)* | t1/2 = 3.4 h | shoulder fat 87% |
| AUC = 15.7 μg h/mL | (10 mpk) fat (90.7 ± 2.6%)* | AUC = 35.6 μg h/mL | Inguinal fat 83% |
| Cl (L/h/kg) = 0.7 | (20 mpk) fat (95.9 ± 0.8%)*** | abdominal cavity fat 81% | |
| F = 68.8% |
Results of in vivo 11β-HSD1 inhibition in rats (?) are expressed as means ± SEMs for n = 4 mice per group. In in vivo 11β-HSD1 inhibition in monkeys, results are expressed as means for n = 2 monkey per group *P < 0.05, and ***P < 0.001 vs vehicle group.
In conclusion, we have developed a series of cyclic sulfamide derivative with an adamantyl group as 11 β-HSD1 inhibitors. Compound 18e showed good in vitro activity toward human and mouse 11β-HSD1, selectivity toward 11β-HSD2, metabolic stability, good PK and safety profiles such as hERG, CYP, and acute toxicity. Additionally, 18e also showed good in vivo efficacy in a primate model.
Supporting Information Available
Synthetic procedures and details of biological assay. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Status
§ These authors contributed equally.
This research was supported by the Center for Biological Modulators of the 21st Century Frontier R&D Program, Ministry of Education, Science and Technology, and by the Ministry of Knowledge Economy (Grant nos. 2011-10033279 and TS113-02).
The authors declare no competing financial interest.
Supplementary Material
References
- Krozowski Z. 11β-Hydroxysteroid dehydrogenase and the short-chain alcohol dehydrogenase (SCAD) superfamily. Mol. Cell. Endocrinol. 1992, 84, C25–C31. [DOI] [PubMed] [Google Scholar]
- Masuzaki H.; Paterson J.; Shinyama H.; Morton N. M.; Mullins J. J.; Seckl J. R.; Flier J. S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [DOI] [PubMed] [Google Scholar]
- Paterson J. M.; Morton N. M.; Fievet C.; Kenyon C. J.; Holmes M. C.; Staels B.; Seckl J. R.; Mullins J. J. Metabolic syndrome without obesity: Hepatic overexpression of 11β-hydroxysteroid dehydrogenase type 1 in trnasgenic mice. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7088–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton N. M.; Paterson J. M.; Masuzaki H.; Holmes M. C.; Staels B.; Fievet C.; Walker B. R.; Flier J. S.; Mullins J. J.; Seckl J. R. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 2004, 53, 931–938. [DOI] [PubMed] [Google Scholar]
- Kotelevtsev Y.; Holmes M. C.; Burchell A.; Houston P. M.; Schmoll D.; Jamieson P.; Best R.; Brown R.; Edwards C. R. W.; Seckl J. R.; Mullins J. J. 11β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 14924–14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge R.; Huang Y.; Liang G.; Li X. 11β-Hydroxysteroid dehydrogenase type 1 inhibitors as promising therapeutic drugs for diabetes: status and development. Curr. Med. Chem. 2010, 17, 412–422. [DOI] [PubMed] [Google Scholar]
- Veniant M. M.; Hale C.; Hungate R. W.; Gahm K.; Emery M. G.; Jona J.; Joseph S.; Adams J.; Hague A.; Moniz G.; Zhang J.; Bartberger M. D.; Li V.; Syed R.; Jordan S.; Komorowski R.; Chen M. M.; Cupples R.; Kim K. W.; St. Jean D. J. Jr.; Johansson L.; Henriksson M. A.; Williams M.; Vallgarda J.; Fotsch C.; Wang M. Discovery of a Potent, Orally Active 11®-Hydroxysteroid Dehydrogenase Type 1 Inhibitor for Clinical Study: Identification of (S)-2-((1S,2S,4R)-Bicyclo[2.2.1]heptan-2-ylamino)-5-isopropyl-5-methylthiazol-4(5H)-one (AMG 221). J. Med. Chem. 2010, 53, 4481–4487. [DOI] [PubMed] [Google Scholar]
- Rosenstock J.; Banarer S.; Fonseca V. A.; Inzucchi S. E.; Sun W.; Yao W.; Hollis G.; Flores R.; Levy R.; Williams W. V.; Seckl J. R.; Huber R. The 11-β-Hydroxysteroid Dehydrogenase Type 1 Inhibitor INCB13739 Improves Hyperglycemia in Patients With Type 2 Diabetes Inadequately Controlled by Metformin Monotherapy. Diabetes Care 2010, 33, 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Hoffman J.; Le P.; Nair S. K.; Cripps S.; Matthews J.; Smith C.; Yang M.; Kupchinsky S.; Dress K.; Edwards M.; Cole B.; Walters E.; Loh C.; Ermolieff J.; Fanjul A.; Bhat G. B.; Herrera J.; Pauly T.; Hosea N.; Paderes G.; Rejto P. The development and SAR of pyrrolidine carboxamide 11β-HSD1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 2897–2902. [DOI] [PubMed] [Google Scholar]
- Tice C. M.; Zhao W.; Xu Z.; Cacatian S. T.; Simpson R. D.; Ye Y.; Singh S. B.; McKeever B. M.; Lindblom P.; Guo J.; Krosky P. M.; Kruk B. A.; Berbaum J.; Harrison R. K.; Johnson J. J.; Bukhtiyarov Y.; Panemangalore R.; Scott B. B.; Zhao Y.; Bruno J. G.; Zhuang L.; McGeehan G. M.; He W.; Claremon D. A. Spirocyclic ureas: Orally bioavailable 11β-HSD1 inhibitors identified by computer-aided drug design. Bioorg. Med. Chem. Lett. 2010, 20, 881–886. [DOI] [PubMed] [Google Scholar]
- Becker C. L.; Engstrom K. M.; Kerdesky F. A.; Tolle J. C.; Wagaw S. H.; Wang W. A Convergent Process for the Preparation of Adamantane 11-β-HSD-1 Inhibitors. Org. Process Res. Dev. 2008, 12, 1114–1118. [Google Scholar]
- Roche D.; Carniato D.; Leriche C.; Lepifre F.; Christmann-Franck S.; Graedler U.; Charon C.; Bozec S.; Doare L.; Schmidlin F.; Lecomte M.; Valeur E. Discovery and structure–activity relationships of pentanedioic acid diamides as potent inhibitors of 11β-hydroxysteroid dehydrogenase type I. Bioorg. Med. Chem. Lett. 2009, 19, 2674–2678. [DOI] [PubMed] [Google Scholar]
- Kwon S. W.; Kang S. K.; Lee J. H.; Bok J. H.; Kim C. H.; Rhee S. D.; Jung W. H.; Kim H. Y.; Bae M. A.; Song J. S.; Ha D. C.; Cheon H. G.; Kim K. Y.; Ahn J. H. Synthesis and 11b hydroxysteroid dehydrogenase 1 inhibition of thiazolidine derivatives with an adamantyl group. Bioorg. Med. Chem. Lett. 2011, 21, 435–439. [DOI] [PubMed] [Google Scholar]
- Maletic M.; Leeman A.; Szymonifka M.; Mundt S. S.; Zokian H. J.; Shah K.; Dragovic J.; Lyons K.; Thieringer R.; Vosatka A. H.; Balkovec J.; Waddell S. T. Bicyclo[2.2.2]octyltriazole inhibitors of 11β-hydoxysteroid dehydrogenase type 1. Pharmacological agents for the treatment of metabolic syndrome. Bioorg. Med. Chem. Lett. 2011, 21, 2568–2572. [DOI] [PubMed] [Google Scholar]
- Sun D.; Wang Z.; Caille S.; DeGraffenreid M.; Gonzales-Lopez de Turiso F.; Hungate R.; Jaen J. C.; Jiang B.; Julian L. D.; Kelly R.; McMinn D. L.; Kaizerman J.; Rew Y.; Sudom A.; Tu H.; Ursu S.; Walker N.; Willcockson M.; Yan X.; Ye Q.; Powers J. P. Synthesis and optimization of novel 4,4-disubstituted cyclohexylbenzamide derivatives as potent 11β-HSD1 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 405–410. [DOI] [PubMed] [Google Scholar]
- Patel J. R.; Shuai Q.; Dinges J.; Winn M.; Pliushchev M.; Fung S.; Monzon K.; Chiou W.; Wang J.; Pan L.; Wagaw S.; Engstrom K.; Kerdesky F. A.; Longenecker K.; Judge R.; Qin W.; Imade H. M.; Stolarik D.; Beno D. W. A.; Brune M.; Chovan L. E.; Sham H. L.; Jacobson P.; Link J. T. Discovery of adamantane ethers as inhibitors of 11β-HSD-1: Synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2007, 17, 750–755. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.