

Abstract
This study demonstrated that cyclomethyline (2) and the corresponding enantiomers (R)-(−)-2 and (S)-(+)-2, displaying α2C-adrenoreceptor (AR) agonism/α2A-AR antagonism, similarly to allyphenyline (1) and its enantiomers, significantly decreased the naloxone-precipitated withdrawal symptoms in mice at very low doses. It also highlighted that such positive effects on morphine dependence can even be improved by additional serotoninergic 5-HT1A receptor (5-HT1A-R) activation. Indeed, 1 or the single (S)-(+)-1, 2, or both its enantiomers, all behaving as α2C-AR agonists/α2A-AR antagonists/5-HT1A-R agonists, alone and at the same low dose, improved morphine withdrawal syndrome and exerted a potent antidepressant-like effect. Therefore, considering the elevated comorbidity between opiate abuse and depressed mood and the benefit of these multifunctional compounds to both disorders, it is possible that they prove more efficacious and less toxic than a cocktail of drugs in managing opioid addiction.
Keywords: α2-adrenergic ligands, 5-HT1A-R agonists, morphine withdrawal symptoms reduction, antidepressant-like effect
Morphine use is the classical approach in the treatment of chronic or cancer-related pain. Nevertheless, even when legally allowed and closely monitored, long-term opioid administration alters central pain-related systems, inducing tolerance and dependence. Opioid addiction, termed a “chronic relapsing disease”, is associated with a myriad of health and social problems; hence, its management is an extremely important area of research.1
α2-Adrenoreceptors (α2-ARs), which have been demonstrated to be extremely sensitive to opioid exposure and to play a key role in opiate withdrawal symptoms,2 belong to the superfamily of G-protein-coupled receptors. The three α2A-, α2B-, and α2C-AR subtypes are widely distributed in the central nervous system (CNS) and peripheral tissues.3 Studies with genetically engineered mice have demonstrated that the α2A subtype mediates hypotension, sedation, and analgesia, as well as inhibition of monoamine release and metabolism in the brain. The α2B subtype mediates vasoconstriction, while the α2C subtype appears to be involved in many CNS processes, such as the startle reflex, stress responses, and control of locomotion as well as feedback inhibition of adrenal cathecolamine release. Moreover, in the brain, α2A- and α2C-ARs, as “heteroreceptors”, inhibit dopamine and serotonin release. Finally, the α2C subtype can contribute to adrenergic-opioid synergy and spinal α2-agonist-mediated analgesia.3−5 In detoxification, α2-AR agonists such as clonidine and lofexidine are often clinically used alone or in combination with traditional treatments to reduce the intensity of withdrawal symptoms, thus increasing treatment duration. However, the α2A-AR activation triggered by these compounds is responsible for side effects such as sedation and hypotension.1 It has been reported that in comparison to clonidine, lofexidine showed decreased incidence and severity of adverse events.1,6 The partial α2A-AR agonism of lofexidine, as recently assessed by us,7 might justify its behavior (Table 1). Therefore, selective α2C-AR agonists, devoid of the side effects associated with α2A-AR stimulation, alone or in combination with opioids, might afford an improvement over current therapies with clonidine-like drugs. We reported the interesting behavior of allyphenyline (1), which displays a significant α2C-AR agonist/α2A-AR antagonist profile8 ascribed to its preferred extended molecular conformation.7 In in vivo studies, 1 enhanced morphine analgesia (due to its α2C-agonism), was devoid of a sedative effect (due to its α2A-antagonism),8,9 and contrasted and prevented morphine tolerance and dependence at a very low dose (0.05 mg/kg).9 In contrast, clonidine attenuated the expression of morphine withdrawal symptoms at a higher dose (5 mg/kg) and was ineffective in preventing them.10 Because α2-AR antagonists (i.e., yohimbine) also attenuated the intensity of withdrawal symptoms,11 we attributed the unique profile of 1 to the synergistic combination of its effective dual α2C-AR agonism/α2A-AR antagonism.9 The positive effects of 1 on dependence were also displayed by (S)-(+)-1 (potent α2C-AR full agonist) and (R)-(−)-1 (α2C-AR partial agonist); they both were endowed with similar α2A-AR antagonism.9 To confirm the aforementioned results and discover novel tools for the management of opioid withdrawal symptoms that have a major role in relapse to drug-taking behavior after detoxification, we extended the study to the recently reported allyphenyline analogue 2,7 now named cyclomethyline, and its enantiomers, prepared in the present investigation. Compound 2 showed interesting α2C-AR agonism/α2A-AR antagonism (Table 1), and according to what reported for 1,7 this functional profile might be associated with a preferred extended molecular conformation. Therefore, the corresponding enantiomers were evaluated by binding and functional assays on Chinese hamster ovary (CHO) cells expressing recombinant human α2-AR subtypes according to previous procedures.9 Moreover, the effects of the racemate 2 at the dose of 0.05 mg/kg and its enantiomers at the dose of 0.025 mg/kg (ip) on the expression and acquisition of morphine dependence were determined. To learn more about the pharmacological properties of this novel class of compounds, expecially in light of the strong comorbidity between opioid addiction and depressive disorders seen in several clinical studies,121 and 2 and their enantiomers were evaluated in a behavioral model of depression in mice. It is known that the serotoninergic [5-hydroxytryptamine (5-HT)] system is involved in the neural regulation of mood, and abnormalities in 5-HT neurotransmission have been observed in the pathophysiology of depression. Moreover, 5-HT dysfunction is one of the main mechanisms contributing to mood disorders in opiate abstinence.13 It has also been suggested that α1A-AR agonists might represent a novel treatment for depression.14 Therefore, the biological profiles of the aforementioned compounds at 5-HT1A receptor (5-HT1A-R), particularly relevant to antidepressant responses in human beings,15 and at α1A-AR were assessed according to procedures previously reported.16 Finally, an assessment of the in vitro absorption, distribution, metabolism, excretion (ADME) properties9 of 2 and of the activity of 1 and 2 on the human ether-à-go-go-related gene (hERG) channel17 was performed.
Table 1. aAffinity (pKi), Antagonist Potency (pKb), Agonist Potency (pEC50), and Intrinsic Activity (ia) on Human α2-AR Subtypesb and Affinity (pKi), Agonist Potency (pD2), and Relative Efficacy (%Emax) on Human 5-HT1A-Rc.
| α2A | α2B |
α2C |
5-HT1A |
||||
|---|---|---|---|---|---|---|---|
| compd | pKb (pKi) | pEC50 (pKi) | ia | pEC50 (pKi) | ia | pD2 (pKi) | %Emax |
| (±)-1 | 7.40 ± 0.06 | NAd | 7.30 ± 0.09 | 0.90 | 6.86 ± 0.09 | 67 | |
| (7.24 ± 0.11) | (6.47 ± 0.20) | (7.07 ± 0.14) | (7.55 ± 0.16) | ||||
| (R)-(−)-1 | 7.40 ± 0.09 | NAd | 6.73 ± 0.11 | 0.50 | <5 | ||
| (7.00 ± 0.08) | (6.25 ± 0.12) | (6.75 ± 0.11) | (<5) | ||||
| (S)-(+)-1 | 7.80 ± 0.13 | 6.00 ± 0.09 | 0.65 | 7.60 ± 0.14 | 0.90 | 7.19 ± 0.10 | 96 |
| (7.28 ± 0.05) | (6.40 ± 0.09) | (7.15 ± 0.09) | (7.45 ± 0.15) | ||||
| (±)-2 | 7.70 ± 0.12 | 5.48 ± 0.15 | 0.70 | 8.70 ± 0.08 | 0.80 | 7.20 ± 0.13 | 75 |
| (7.44 ± 0.09) | (6.39 ± 0.05) | (6.56 ± 0.21) | (7.98 ± 0.07) | ||||
| (R)-(−)-2 | 7.20 ± 0.11 | 5.40 ± 0.11 | 0.65 | 6.50 ± 0.07 | 0.75 | 6.80 ± 0.10 | 68 |
| (7.35 ± 0.10) | (6.47 ± 0.07) | (6.63 ± 0.05) | (7.40 ± 0.08) | ||||
| (S)-(+)-2 | 7.70 ± 0.09 | 5.80 ± 0.06 | 0.70 | 8.70 ± 0.11 | 0.85 | 7.40 ± 0.02 | 97 |
| (7.38 ± 0.07) | (6.39 ± 0.10) | (6.56 ± 0.13) | (8.24 ± 0.16) | ||||
| clonidineb | e | 5.95 ± 0.27 | 0.45 | 7.24 ± 0.14 | 0.60 | ||
| (5.49 ± 0.13) | |||||||
| lofexidinef | g | 6.90 ± 0.14 | 0.80 | 8.86 ± 0.20 | 0.80 | ||
| (8.36 ± 0.11) | (7.17 ± 0.18) | (7.16 ± 0.07) | (6.90 ± 0.11) | ||||
| 8-OH-DPAT | 7.60 ± 0.06 | 100 | |||||
| (8.47 ± 0.13) | |||||||
The data were expressed as means ± SEMs of 3–6 separate experiments.
According to ref (9).
pKi values were derived from displacement of [3H]-8-OH-DPAT binding on human cloned 5-HT1A-R. pD2 values are the negative logarithm of the agonist concentration required to obtain 50% of the maximal stimulation of [35S]GTPγS binding. Maximal stimulation (%Emax) is expressed as a percentage of the maximal 8-OH-DPAT response.
Compounds exhibiting ia of <0.3 were considered not active (NA).
pEC50 = 8.08 ± 0.12; ia = 0.90.
Ref (7).
pEC50 = 8.20 ± 0.10; ia = 0.60.
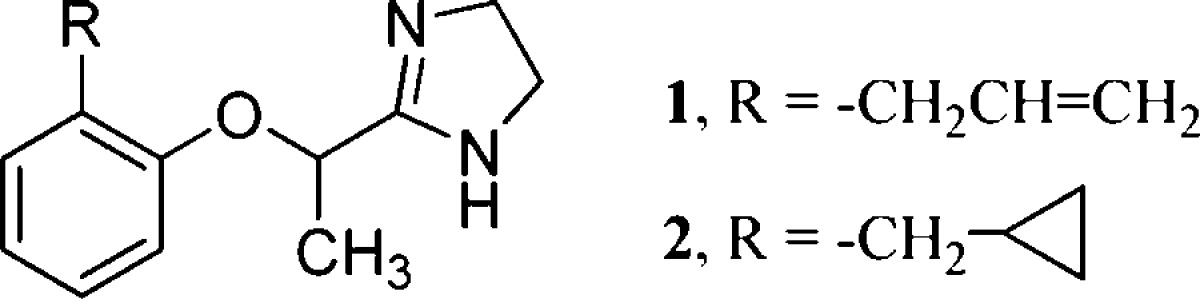
The enantiomers of 2 were prepared according to Scheme 1. (S)-(+)- and (R)-(−)-2-(2-cyclopropylmethyl)propionic acid methyl ester [(S)-(+)-3 and (R)-(−)-3, respectively] were obtained by Mitsunobu reaction from 2-(cyclopropylmethyl)phenol18 and methyl (R)-(−)- or (S)-(+)-lactate, respectively, with inversion of configuration.19 The reaction of (S)-(+)- or (R)-(−)-3 with ethylenediamine in the presence of (CH3)3Al yielded the corresponding imidazolines (S)-(+)- and (R)-(−)-2, whose e.e. determined by 1H NMR spectroscopy was about 85%, due to partial racemization occurring under the conditions used in the reactions. Indeed, their spectra, as well as that of the racemic (±)-2 on addition of the chiral shift reagent (S)-(+)-2,2,2-trifluoro-1-(9-anthryl)-ethanol, showed a double doublet at δ 1.38 ppm for the methyl protons. The resolution of the racemic imidazoline (±)-2 was performed by HPLC using Chiralcel OD-H (25 cm × 0.46 cm, 5 μm particle size) as the chiral stationary phase and n-hexane/2-propanol 90/10 v/v as the mobile phase. Detection was monitored at a wavelength of 220 nm, and the retention times were 5.9 and 10.3 min for (+)-2 and (−)-2, respectively; (+)-2: [α]D20 = +19.28, e.e. 100%; (−)-2: [α]D20 = −19.59, e.e. 100%. In this case, the 1H NMR spectra, similarly performed, showed for the methyl group of (+)-2 and (−)-2 only one doublet at δ 1.36 and δ 1.40 ppm, respectively. The absolute configuration of the stereocenter in the bridge of these enantiomers was determined by comparing the sign of their optical rotations with those of the enantiomers obtained by stereoselective synthesis. Consequently, the (S) absolute configuration was assigned to the dextrorotatory enantiomer (+)-2 and (R) to the levorotatory (−)-2.
Scheme 1. Synthesis of (S)-(+)-2 and (R)-(−)-2.

Reagents and conditions: (a) DIAD, Ph3P, THF, rt. (b) Ethylenediamine, Al(CH3)3, toluene, 0–65 °C, 7 h.
The affinity values (pKi), antagonist properties (pKb), agonist potencies, and intrinsic activities (pEC50 and ia, respectively) of the enantiomers of 2 on α2-AR subtypes are reported in Table 1. The biological profiles of 2, 1, and its enantiomers were also included for useful comparison. This study indicates that (i) both enantiomers of 2 displayed binding affinities for the three α2-AR subtypes comparable to those of the racemic compound; (ii) (S)-(+)-2 was endowed with the highest α2C-AR agonist activity; (iii) (R)-(−)-2 was characterized by lower but still significant α2C-AR agonism; and (iv) (R)-(−)-2 and (S)-(+)-2 showed weak α2B-AR activation but effective α2A-AR antagonist potencies. Interestingly, 2, (R)-(−)-2 and (S)-(+)-2 affected expression and acquisition of morphine dependence (Figure 1) as observed previously for 1 and its enantiomers.9 The frequencies of naloxone-precipitated jumping and other somatic signs (rearing, forepaw tremors, and teeth chatter) decreased significantly. To study possible antidepressant-like activity, the mouse forced swimming test (FST), a behavioral model with good reliability and predictive validity for screening antidepressants,20 was used. Figure 2A shows that 1 induced significant reduction of immobility time. This effect was maximal at the dose of 0.05 mg/kg and comparable to that obtained with the dose of 20 mg/kg of the selective 5-HT reuptake inhibitor fluoxetine, included as reference compound.21 Significant antidepressant activity was also observed at 0.025 and 0.2 mg/kg doses, whereas no activity was observed at lower and higher doses. Similarly, the maximal effect of 2 was displayed at the dose of 0.05 mg/kg (Figure 2B). Nevertheless, the antidepressant response of 1 was only associated with the (S)-(+)-1 enantiomer, whereas the effect of 2 was similarly induced by both its enantiomers (Figure 3). In Table 1, affinities and functional values of all compounds at 5-HT1A-R are also reported. The 5-HT1A-R agonist 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) was included for useful comparison.16 From the data, it emerged that 1 displayed good 5-HT1A-R affinity and behaved as an agonist. These properties were maintained and even enhanced only by the (S)-(+)-1 enantiomer, whereas (R)-(−)-1 showed negligible affinity (pKi < 5).
Figure 1.

Effects of acute (A, B) or repeated (C, D) ip administration of (±)-2 (0.05 mg/kg), (R)-(−)-2, and (S)-(+)-2 (0.025 mg/kg) on expression and acquisition of morphine dependence, respectively. Naloxone-precipitated withdrawal symptoms, in both control and morphine-treated mice, are given as a measure of the frequency of jumping (A, C) and somatic signs (B, D) expressed as a summary of rearing, forepaw tremors, and teeth chatter. Significant differences: *p < 0.05, **p < 0.01, as compared to vehicle; ++p < 0.01, as compared to the morphine group.
Figure 2.
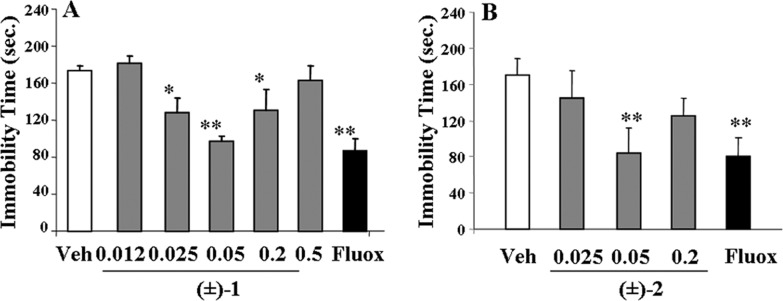
Immobility time in the FST in mice following ip administration of (±)-1 at 0.012, 0.025, 0.05, 0.2, and 0.5 mg/kg (A), (±)-2 at 0.025, 0.05, and 0.2 mg/kg (B), and fluoxetine (Fluox; 20 mg/kg) as a reference drug. Data represent means (±SEMs) of 8–9 animals. Differences from vehicle-treated: *p < 0.05, **p < 0.01; where not indicated, the differences are not statistically significant.
Figure 3.
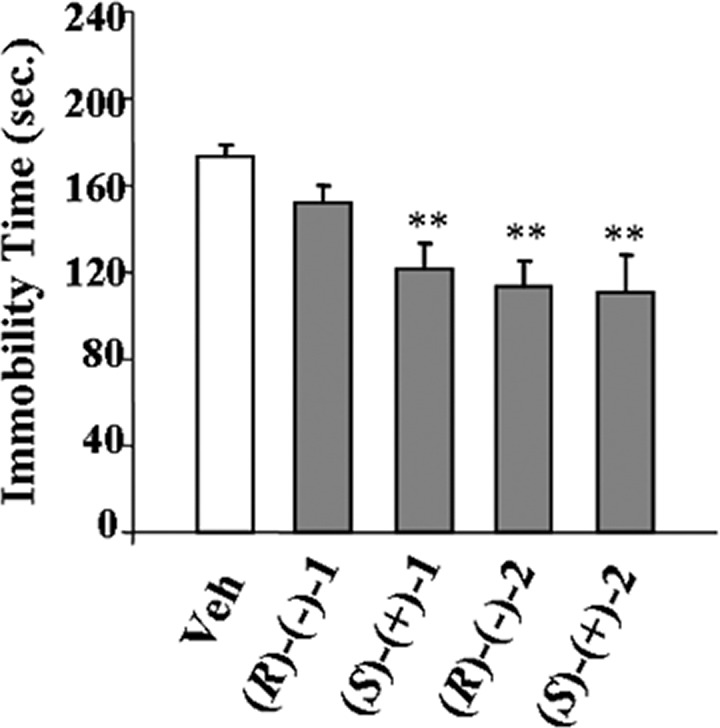
Immobility time in the FST in mice following ip administration of (R)-(−)-1, (S)-(+)-1, (R)-(−)-2, and (S)-(+)-2 (0.025 mg/kg). Data represent means (±SEMs) of eight animals. Differences from vehicle-treated: **p < 0.01; where not indicated, the differences are not statistically significant.
In contrast, 2, (R)-(−)-2 and (S)-(+)-2 displayed similar and significant 5-HT1A-R affinities and agonist potencies. None of the compounds studied showed significant α1A-AR agonism (ia < 0.35). Altogether, the data indicated that dual α2C-AR/5-HT1A-R activation was requested for the antidepressant-like effect induced by low doses of 1 and the single (S)-(+)-1 enantiomer, 2, and both its enantiomers. This consideration does not contrast with the results of previous studies.22,23 Indeed, they indicated that low doses of clonidine (0.01–0.06 mg/kg) increased 5-HT neurotransmission by attenuating the release of endogenous noradrenaline via activation of α2-AR autoreceptors on adrenergic neurones22 and, hence, induced anti-immobility effects with subactive doses of 5-HT1A-R agonists (i.e., 8-OH-DPAT) in mouse FST.23 On the contrary, higher doses (0.1–0.4 mg/kg) decreased 5-HT neurotransmission through the direct activation of the α2-AR heteroreceptors on the 5-HT terminals.22 The peculiar relationship between α2-AR activation and 5-HT function, highlighted by such studies, might justify the dose-dependent U-shaped trend exhibited by the anti-immobility effect of our α2C-AR/5-HT1A-R agonists. To support our observations, experiments in the presence of the 5-HT1A-R antagonist WAY 10013524 and the α2-AR antagonist yohimbine25 were carried out. Both antagonists, ineffective when given alone, significantly and similarly contrasted the actions evoked by 0.025 mg/kg dose of (S)-(+)-1, (R)-(−)-2, and (S)-(+)-2 (Figure 4). Therefore, these results confirmed that dual α2C-AR/5-HT1A-R activation, favorably triggered by the same molecule, took part in the anti-immobility effect. It has been reported that selective α2C-AR antagonists, allowing α2A-AR stimulation, produced important antidepressant effects in the FST.26 Because α2A- and α2C-AR subtypes complement each other to integrate CNS function and behavior,5 it is possible that also the α2C-AR agonism/α2A-AR antagonism combination, associated with 5-HT1A activation, as verified for the examined compounds, is equally compatible with an antidepressant-like effect. The advantage of this peculiar biological profile was also supported by the observation that yohimbine, a 5-HT1A-R agonist27 and antagonist at all three α2-AR subtypes,25 did not display antidepressant activity. The physicochemical and in vitro ADME9 profile of 2 proved to be similar to that of 1. Indeed, 2 was highly soluble in water at physiological pH. The permeation rate in a parallel artificial membrane permeability assay (PAMPA) assay and the percentage remaining after incubation for 1 h with recombinant hCYP3A4 were very high. From an automatic patch clamp assay on HEK293 cells overexpressing the hERG channel,17 compounds 1 and 2 showed to be weak inhibitors, with IC50 values of 16.9 and 8.9 μM, respectively, suggesting a low cardiotoxicity risk associated with their potential therapeutic use.
Figure 4.
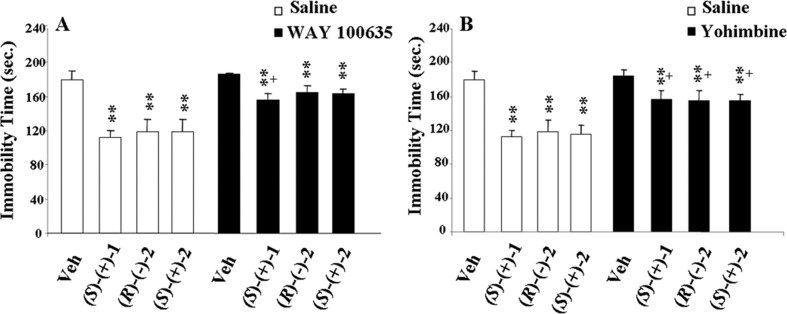
Influence of the 5-HT1A-R antagonist WAY 100635 (0.1 mg/kg, sc) (A) and the α2-AR antagonist yohimbine (1.250 mg/kg, ip) (B) on the antidepressant-like effect of the enantiomers (S)-(+)-1, (R)-(−)-2, and (S)-(+)-2 (0.025 mg/kg, ip) on the FST. Data represent means (±SEMs) of 7–8 animals. Significant differences: **p < 0.01, as compared with vehicle group; ++p < 0.01, as compared with enantiomers related-treated mice; and °p < 0.05, as compared with antagonist-treated mice; where not indicated, the differences are not statistically significant.
In summary, the present study demonstrated that cyclomethyline (2) and its enantiomers, characterized by α2C-AR agonist/α2A-AR antagonist profile, significantly decreased the naloxone-precipitated withdrawal symptoms in mice at very low doses. These properties are in agreement with those shown by the lead 1 and its enantiomers and confirm that dual α2C-AR agonism/α2A-AR antagonism represents a favorable condition for inducing positive effects on morphine dependence.9 Interestingly, such effects can even be improved by additional 5-HT1A-R activation. Indeed, unlike clonidine, 1 or the single (S)-(+)-1 enantiomer, 2, or both its enantiomers, all behaving as α2C-AR agonists/α2A-AR antagonists/5-HT1A-R agonists, alone and at the same low dose, improved morphine withdrawal syndrome and exerted a potent antidepressant-like effect. It has been suggested that opiate abuse may induce depressed mood, or conversely, depression can drive to drug abuse in a self-medication hypothesis. Therefore, such multifunctional compounds might be beneficial to both disorders.13 In addition, their therapeutic potential is strongly enhanced by the lack of sedative side effects (due to their α2A-antagonism) and alteration of motor activity (data not shown), as well as by their favorable in vitro ADME profiles and limited activity on the hERG channel.
Acknowledgments
We thank the MIUR (Rome), the University of Camerino, and the Fondazione Monte dei Paschi (Siena) for financial support.
Glossary
Abbreviations
- AR
adrenoreceptor
- CNS
central nervous system
- CHO
Chinese hamster ovary
- 5-HT
5-hydroxytryptamine
- 5-HT1A-R
5-HT1A receptor
- ee
enantiomeric excess
- ip
intraperitoneally
- sc
subcutaneously
- ADME
absorption, distribution, metabolism, excretion
- PAMPA
parallel artificial membrane permeability assay
- DIAD
diisopropyl azodicarboxylate
- CYP
cytochrome P450
- hERG
human ether-à-go-go-related gene
Supporting Information Available
Synthetic procedure, hERG activity assay, detail of biological assay, and elemental analysis of the final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Veilleux J. C.; Colvin P. J.; Anderson J.; York C.; Heinz A. J. A review of opioid dependence treatment: pharmacological and psychological interventions to treat opioid addiction. Clin. Psychol. Rev. 2010, 30, 155–166and references therein. [DOI] [PubMed] [Google Scholar]
- Streel E.; Dan B.; Campanella S.; Meyvaert A.; Hanak C.; Pelc I.; Verbanck P. A pharmacological modulation of opiate withdrawal using an up-/down-regulation of the noradrenergic system in opiate-dependent rats. J. Neuropsychoparmacol. 2006, 9, 621–626. [DOI] [PubMed] [Google Scholar]
- Tan C. M.; Limbird L. E.. The α2-adrenergic receptors. In The Receptors: The Adrenergic Receptors in the 21st Century; Perez D., Ed.; Humana Press Inc.: Totowa, NJ, 2006; pp 241–265. [Google Scholar]
- Fairbanks C. A.; Stone L. S.; Wilcox G. L. Pharmacological profile of alpha 2 adrenergic receptor agonists identified using genetically altered mice and isobolographic analysis. Pharmacol. Ther. 2009, 123, 224–238and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein L. Adrenoceptors and signal transduction in neurons. Cell Tissue Res. 2006, 326, 541–551and references therein. [DOI] [PubMed] [Google Scholar]
- Gish E. C.; Miller J. L.; Honey B. L.; Johnson P. N. Lofexidine, an alpha2-receptor agonist for opioid detoxification. Ann. Pharmacother. 2010, 44, 343–351. [DOI] [PubMed] [Google Scholar]
- Diamanti E.; Del Bello F.; Carbonara G.; Carrieri A.; Fracchiolla G.; Giannella M.; Mammoli V.; Piergentili A.; Pohjanoksa K.; Quaglia W.; Scheinin M.; Pigini M. Might the observed α2A-adrenoreceptor agonism or antagonism of allyphenyline analogues be ascribed to different molecular conformations?. Bioorg. Med. Chem. 2012, 20, 2082–2090. [DOI] [PubMed] [Google Scholar]
- Cardinaletti C.; Mattioli L.; Ghelfi F.; Del Bello F.; Giannella M.; Bruzzone A.; Paris H.; Perfumi M.; Piergentili A.; Quaglia W.; Pigini M. Might adrenergic α2C-agonists/α2Aantagonists become novel therapeutic tools for pain treatment with morphine?. J. Med. Chem. 2009, 52, 7319–7322and references therein. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Mattioli L.; Ghelfi F.; Giannella M.; Piergentili A.; Quaglia W.; Cardinaletti C.; Perfumi M.; Thomas R. J.; Zanelli U.; Marchioro C.; Dal Cin M.; Pigini M. Fruitful adrenergic α2C-agonism/α2A-antagonism combination to prevent and contrast morphine tolerance and dependence. J. Med. Chem. 2010, 53, 7825–7835and references therein. [DOI] [PubMed] [Google Scholar]
- Özdoğan Ü. K.; Lähdesmäki J.; Scheinin M. Influence of prazosin and clonidine on morphine analgesia, tolerance and withdrawal in mice. Eur. J. Pharmacol. 2003, 460, 127–134. [DOI] [PubMed] [Google Scholar]
- Özdogan Ü, K.; Lähdesmäki J.; Hakala K.; Scheinin M. The involvement of α2A-adrenoceptors in morphine analgesia, tolerance and withdrawal in mice. Eur. J. Pharmacol. 2004, 497, 161–171. [DOI] [PubMed] [Google Scholar]
- Cerdá M.; Sagdeo A.; Galea S. Comorbid forms of psychopathology: key patterns and future research directions. Epidemiol. Rev. 2008, 30, 155–177. [DOI] [PubMed] [Google Scholar]
- Goeldner C.; Lutz P.-E.; Darcq E.; Halter T.; Clesse D.; Ouagazzal A.-M.; Kieffer B. L. Impaired emozional-like behavior and serotonergic function during protracted abstinence from chronic morphine. Biol. Psychiatry 2010, 69, 236–244and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doze V. A.; Handel E. M.; Jensen K. A.; Darsie B.; Luger E. J.; Haselton J. R.; Talbot J. N.; Rorabaugh B. R. α1A- and α1B-adrenergic receptors differentially modulate antidepressant-like behaviour in the mouse. Brain Res. 2009, 1285, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blier P.; Ward N. M. Is there a role for 5-HT1A agonists in the treatment of depression?. Biol. Psychiatry 2003, 53, 193–203. [DOI] [PubMed] [Google Scholar]
- Quaglia W.; Piergentili A.; Del Bello F.; Farande Y.; Giannella M.; Pigini M.; Rafaiani G.; Carrieri A.; Amantini C.; Lucciarini R.; Santoni G.; Poggesi E.; Leonardi A. Structure-activity relationships in 1,4-benzodioxan-related compounds. From 1,4-benzodioxan to 1,4-dioxane ring as a promising template of novel α1D-adrenoreceptor antagonists, 5-HT1A full agonists, and cytotoxic agents. J. Med. Chem. 2008, 51, 6359–6370. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M. C.; Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [DOI] [PubMed] [Google Scholar]
- He Z.; Yudin A. K. Palladium-catalyzed oxidative activation of arylcyclopropanes. Org. Lett. 2006, 25, 5829–5832. [DOI] [PubMed] [Google Scholar]
- Tottie L.; Baeckstrom P.; Moberg C.; Tegenfeldt J.; Heumann A. Molecular sieve controlled diastereoselectivity: Effect in the palladium-catalyzed cyclization of cis-1,2-divinylcyclohexane with R-oxygen-substituted acids as chiral nucleophiles. J. Org. Chem. 1992, 57, 6579–6587. [Google Scholar]
- Petit-Demouliere B.; Chenu F.; Bourin M. Forced swimming test in mice: A review of antidepressant activity. Psychopharmacology 2005, 177, 245–255. [DOI] [PubMed] [Google Scholar]
- Page M. E.; Detke M. J.; Dalvi A.; Kirby L. G.; Lucki I. Serotonergic mediation of the effects of fluoxetine, but not desipramine, in the rat forced swimming test. Psychopharmacology 1999, 147, 162–167. [DOI] [PubMed] [Google Scholar]
- Mongeau R.; Blier P.; de Montigny C. In vivo electrophysiological evidence for tonic activation by endogenous noradrenaline of alpha 2-adrenoceptors on 5-hydroxytryptamine terminals in the rat hippocampus. Naunyn-Schmiedebergs Arch. Pharmacol. 1993, 347, 266–272. [DOI] [PubMed] [Google Scholar]
- Redrobe J. P.; Bourin M. Clonidine potentiates the effects of 5-HT1A, 5-HT1B and 5-HT2A/2C antagonists and 8-OH-DPAT in the mouse forced swimming test. Eur. J. Neuropsychofarmacol. 1998, 8, 169–173. [DOI] [PubMed] [Google Scholar]
- Cliffe I. A.; Brightwell C. I.; Fletcher A.; Forster E. A.; Mansell H. L.; Reilly Y.; Routledge C.; White A. C. (S)-N-tert-Butyl-3-(4-(2-methoxyphenyl)piperazin-1-yl)-2-phenylpropanamide [(S)-WAY-100135]: A selective antagonist at presynaptic and postsynaptic 5-HT1A receptors. J. Med. Chem. 1993, 36, 1509–1510. [DOI] [PubMed] [Google Scholar]
- Audinot V.; Fabry N.; Nicolas J.-P.; Beauverger P.; Newman-Tancredi A.; Millan M. J.; Try A.; Bornancin F.; Canet E.; Boutin J. A. Ligand modulation of [35S]GTPγS binding at human α2A, α2B and α2C adrenoceptors. Cell Signalling 2002, 14, 829–837. [DOI] [PubMed] [Google Scholar]
- Sallinen J.; Höglund I.; Engström M.; Lehtimäki J.; Virtanen R; Sirviö J.; Wurster S.; Savola J.-M.; Haapalinna A. Pharmacological characterization and CNS effects of a novel highly selective α2C-adrenoceptor antagonist JP-1302. Br. J. Pharmacol. 2007, 150, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A.; Quirk G. J. Pharmacological facilitation of fear extinction and the search for adjunct treatments for anxiety disorders—The case of yohimbine. Trends Pharmacol. Sci. 2009, 31, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.