

Abstract
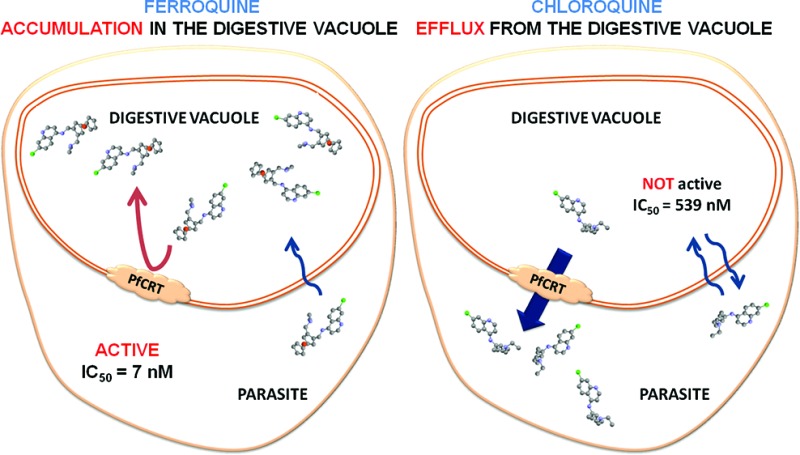
The aminoquinoline chloroquine (CQ) has been widely used for treating malaria since World War II. Resistance to CQ began to spread around 1957 and is now found in all malarious areas of the world. CQ resistance is caused by multiple mutations in the Plasmodium falciparum chloroquine resistance transporter (PfCRT). These mutations result in an increased efflux of CQ from the acidic digestive vacuole (DV) to the cytosol of the parasite. This year, we proposed a strategy to locate and quantify the aminoquinolines in situ within infected red blood cells (iRBCs) using synchrotron based X-ray nanoprobe fluorescence. Direct measurements of unlabeled CQ and ferroquine (FQ) (a ferrocene-CQ conjugate, extremely active against CQ-resistant strains) enabled us to evidence fundamentally different transport mechanisms from the cytosol to the DV between CQ and FQ in the CQ-susceptible strain HB3. These results inspired the present study of the localization of CQ and FQ in the CQ-resistant strain W2. The introduction of the ferrocene core in the lateral side chain of CQ has an important consequence: the transporter is unable to efflux FQ from the DV. We also found that resistant parasites treated by FQ accumulate a sulfur-containing compound, credibly glutathion, in their DV.
Keywords: drug delivery, drug resistance, antiparasitic agents, ferroquine, chloroquine
Despite progress in fighting malaria, this infectious disease causes 700000 deaths per year worldwide, mostly in children under five.1 The 4-aminoquinoline chloroquine (CQ) (Figure 1) was used extensively after World War II for malaria prevention and treatment. Approximately 50 years ago, the hope inspired by the success of CQ was destroyed due to the emergence of resistant parasites.2 Thus, it is essential to constantly develop new antimalarials against Plasmodium falciparum.3 To address this need, in the mid-1990s, we initiated a structure-based drug design program wherein ferrocenyl analogues of antimalarial drugs in current use were synthesized and tested. Ferroquine (FQ or SSR97193, Figure 1) emerged from this project as a novel antimalarial drug candidate that completed phase 2 clinical trials in combination with artesunate for the treatment of uncomplicated malaria.4 Recently, we proposed a novel strategy to locate and quantify drugs in pharmacological doses in vitro with sufficient spatial resolution and without labeling.5
Figure 1.

Chemical structures of CQ and FQ and their antimalarial activity against the W2 strain of P. falciparum in vitro.
These results inspired the present study of the localization of CQ and FQ in the CQ-resistant P. falciparum strain W2. Indeed, even if the mechanism of action of both drugs is linked with the heme detoxification pathway in the digestive vacuole (DV) of the parasite, we hypothesized different molecular modes of action in susceptible and resistant parasites.
The pioneering work of Sullivan and Golberg6 showed that CQ-resistant parasites accumulate significantly less CQ in their DV than CQ-susceptible parasites. After this discovery, numerous studies were reported, speculating on the amount of CQ in CQ-resistant versus CQ-susceptible parasites.7
Unfortunately, the absence of standardized experimental protocols led to a variety of data depending on the strains, the external concentration of CQ, the incubation time, etc. Nevertheless, the general assumption is that CQ-resistant parasites accumulate 2–10-fold less CQ than CQ-susceptible at pharmacological doses (1–50 nM)8−10 with the most probable explanation being an increased efflux of CQ out of the DV.9 Using purified P. falciparum chloroquine resistance transporter (PfCRT), kinetic data on quinoline transport by PfCRT showed little change in the rate constants for mutant versus wild-type proteins.11 Polymorphims in PfCRT, particularly in the K76T mutation, have been demonstrated to bring about CQ resistance.12 The function of PfCRT as a channel or a carrier is still an open question.13 Martin et al. have published evidence that PfCRT behaves as a carrier.14 However, data from purified proteoliposomes containing purified PfCRT suggest a channel-like mechanism.11 In wild-type PfCRT, the protonated lysine could repulse the (di)protonated forms of CQ, thereby preventing its transport and maintaining a lethal concentration in the DV, whereas in mutated PfCRT, mutation of lysine 76 to threonine could allow interaction between PfCRT and charged CQ and therefore allos its efflux out of the DV.15,16 Recently, Roepe's group showed that Saccharomyces cerevisiae yeast expressing either CQ-susceptible or CQ-resistant isoforms of PfCRT are both capable of increased CQ transport.17 Moreover, resistant PfCRT leads to increased transport.17 The majority of available transport data and drug binding studies18,19 suggest that both isoforms interact with and transport the drug. Other genes known to be associated with quinoline resistance are pfmdr1, pfmrp, and pfnhe-1.
The strain of P. falciparum used for demonstrating the in vitro differential accumulation between FQ and CQ was W2, an Indochinese clone exhibiting CQ resistance. To study the subcellular distribution FQ and CQ within infected red blood cells (iRBCs), we used a synchrotron-based X-ray fluorescence nanoprobe. As a diprotic weak base with pKa values of 7.00 and 8.45, FQ can exist in a neutral, monoprotonated, or diprotonated form under the acidic DV environment.20 As with CQ,21 these species should have different reactivities toward the different forms of heme (or hematin). In this study, we only used the unlabeled drugs CQ and FQ to avoid any artifact that could lead to wrong conclusions.20 Indeed, if the physicochemical properties of ruthenoquine, the cold tracer analogue of FQ, are similar to those of FQ, they are not identical. For example, RQ, with pKa values of 6.99 and 7.97, is less basic than FQ. Nevertheless, the idea of using the chlorine atom of the drugs asa labeling agent avoids any quantification of the drugs due to the presence of a large amount of Cl ions under physiological conditions and/or coming from the buffer.
Experiments were performed on W2 strains exposed for 30 min to 40 nM FQ (or CQ). The European Synchrotron Radiation Facility nanoimaging end station can achieve 50–100 nm X-ray spot range at 25 keV excitation with very high flux.22 Details on the preparation of the P. falciparum iRBCs and the X-ray fluorescence are given in the Experimental Section. The area of high iron content corresponded to hemozoin (Hz) crystals (Figure 2) and perfectly matched the presence of the malaria pigment. In untreated iRBCs (control), homogeneous Cl signals were recorded in the whole sample (RBC, parasite, and DV) corresponding to background noise of residual Cl ions (Figure 2, control). The same remark can be made for the S signals (Figure 2, control). In iRBCs treated with FQ, the area of high Cl content corresponded to the DV (Figure 2). The colocalization of Cl and Fe atoms with Hz in the DV strongly suggests the presence of FQ within this compartment in CQ-resistant strain. To the contrary, in iRBCs treated with CQ, the area of DV corresponds to a chlorine-depleted area. This also contrasts to our previous results on CQ in the DV of CQ-sensitive strain.5 The highest Cl signal was measured in the area of the iRBC excluding the DV. The Cl/Fe ratio was derived from calculated mass fraction within the DV area and was found to be significantly higher for FQ as compared to CQ-treated W2 cells within our experimental conditions (2.4 ± 0.3 vs 1.6 ± 0.5 respectively, N = 4). While spatial resolution is still limited using synchrotron-based X-ray fluorescence nanoprobe (50–100 nm), we can conclude that CQ-resistant parasites accumulate FQ, whereas efflux of CQ out of the DV takes place.
Figure 2.

Intraerythrocytic distribution of the antimalarial drugs FQ and CQ in the W2 strain of P. falciparum in vitro. Iron (Fe), chlorine (Cl), and sulfur (S) Kα X-ray fluorescence intensity maps for control iRBCs and iRBCs exposed for 30 min to CQ and FQ (40 nM) (see ESI for further details). The optical micrograph of P. falciparum iRBC is added for clarity.
In 2008, Pradines' group hypothesized that FQ may not interact with transport proteins (Pfcrt, Pfmdr1, Pfmrp, and Pfnhe-1) in quinoline-resistant parasites.23 Coupling these observations altogether, it is clear that Pfcrt, Pfmdr1, Pfmrp, and Pfnhe-1 are unable to transport FQ out of the DV, thus allowing FQ to evade the resistance mechanisms.
During the oxidative process of hemoglobin digestion, the parasite is exposed to high fluxes of reactive oxygen species (ROS). Glutathione (GSH), a tripeptide of glutamic acid, cysteine, and glycine are known to protect P. falciparum from oxidative damage.24,25 As compared with untreated iRBCs, the area of high sulfur content in iRBCs treated with FQ corresponds to the DV (Figure 2). The colocalization of S and Fe atoms in the DV indicates the accumulation of a sulfur-containing compound within this organelle. To the contrary, in iRBCs treated with CQ, the area of the DV corresponds to a depleted S content within the DV. For comparison, Figure 3 shows the differences between CQ and FQ DV accumulation for CQ-susceptible HB35 versus CQ-resistant W2 parasites. The mass fraction could be calculated, and the derived S/Fe ratio is found significantly higher for FQ as compared to CQ-treated W2 cells (1.8 ± 0.3 vs 1.2 ± 0.3, respectively, N = 4). What is the nature of the sulfur-containing compound accumulated in the DV? While we cannot exclude the presence of a sulfur-containing protein,26 the most simple and rational hypothesis is that this subcellular accumulation is due to a higher concentration in GSH in iRBCs treated with FQ (as a response to oxidative aggression) than in iRBCs treated with CQ (with an absence of oxidative aggression). As previously hypothesized, FQ should be able to enhance the oxidative stress in the acidic and oxidative conditions of the DV.16 Two pathways could therefore be envisaged; FQ in the presence of H2O2 generates OH• radicals via a Fenton-like reaction, causing damage to the membranes or the proteins of the parasite DV; or FQ inhibits the formation of the malaria parasite or disrupts the hemozoin. Accumulation of toxic heme (free or complexed with FQ) leads to the death of malaria parasite.
Figure 3.
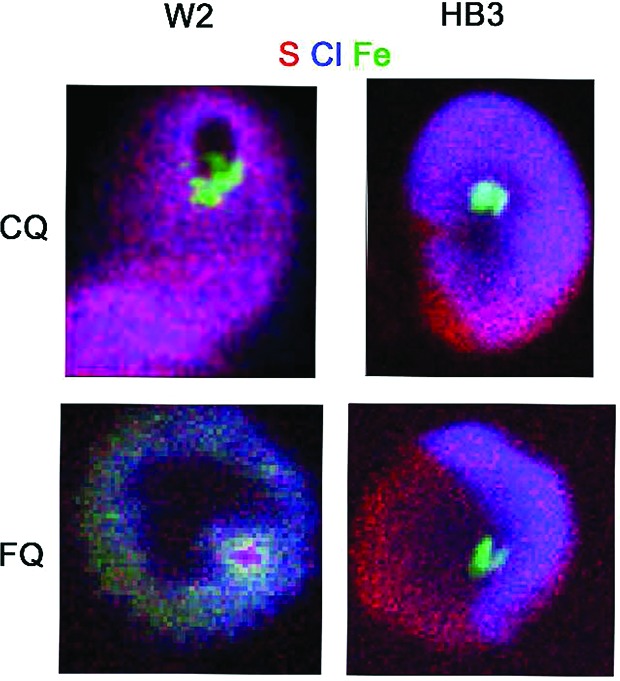
Comparison of the intraerythrocytic distribution of the antimalarial drugs FQ and CQ in the HB3 and W2 strains of P. falciparum in vitro. After overlaying the sulfur (S, red), iron (Fe, green), and chlorine (Cl, blue) maps, the colocalization of Cl and Fe appeared in light blue. HB3 maps are reproduced from ref (5). Copyright 2012 The Royal Society of Chemistry.
Our observation of sulfur accumulation in the DV of CQ-resistant parasites treated by FQ provides a strong argument for the generation of oxidative stress by FQ and for a defense response probably based on GSH, which is an efficient trapping molecule for OH•. As CQ is unable to generate oxidative stress and does not accumulate in the DV of the W2 strain, such a sulfur accumulation is not observed (Figure 3). Remarkably, these unexpected findings of a large amount of sulfur in the DV validate recent results of Patzewitz et al. on the essential role of GSH for Plasmodium.27 These authors suggest that mutant PfCRT transport GSH into the DV for resistant parasites.
The feasibility of comparing the distribution of FQ and CQ in CQ-resistant P. falciparum by a synchrotron X-ray nanoprobe has been demonstrated. We were able to locate FQ in the DV, while CQ is effluxed out the DV of the parasite. Mapping the S content led us to conclude that the increase of the oxidative stress in the DV of the parasites treated by FQ, but not CQ, is a more and more relevant hypothesis.
The FQ conundrum illustrates the quote of Martin “Quinolines are not passé”.28 The question “why does the introduction of a ferrocene moiety in an old drug reduce the induction of resistance?” has still not yet been answered. Nevertheless, there are a number of answers: (1) FQ is not recognized and transported out of the DV by PfCRT or other proteins involved in quinoline-resistance, (2) the physicochemical properties of FQ result in accumulation within the DV where FQ is a more potent inhibitor of hemozoin formation than CQ, and (3) the ferrocene core induces directly or indirectly ROS generation within the DV (Figure 4). Taken altogether, these data require a new theory for the mechanism of antimalarial action of FQ.
Figure 4.
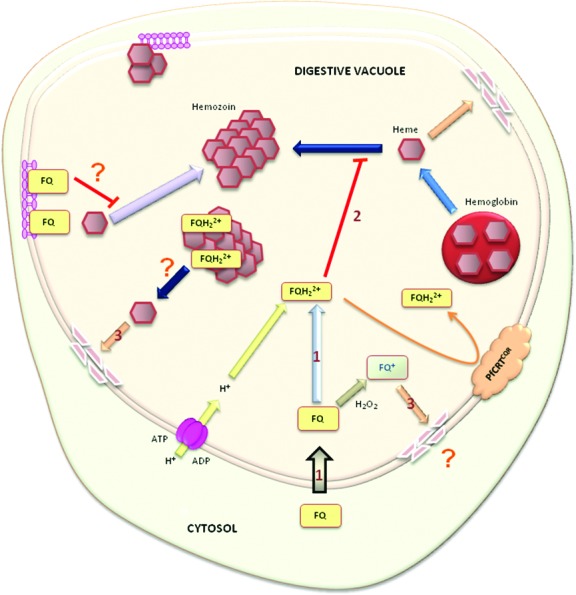
Hypothetical models for the mechanism of action of FQ. (1) FQ accumulates within the DV through passive diffusion and/or through an active transporter. The intramolecular H-bond formed in FQ improves membrane permeability. (2) FQ opens up and inhibits the formation of the malaria pigment by interacting with the {001} or {100} crystal faces. (3) In the oxidizing conditions of the DV, FQ should be capable of undergoing redox reactions causing lipid oxidation. Interactions between malaria pigment and FQs destabilize hemozoin and liberate hematin (free or complexed with FQ), leading to membrane damage. The interactions of FQ with membrane lipids are also supposed to inhibit the crystallization of hemozoin. The K76T polymorphisms in PfCRT do not confer resistance to FQ. A lethal concentration of FQ is maintained in the DV even in quinoline-resistant parasites.
Acknowledgments
We are grateful to Hadidjatou Kalamou for technical support. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities.
Glossary
Abbreviations
- CQ
chloroquine
- FQ
ferroquine
- iRBCs
infected red blood cells
- PfCRT
P. falciparum chloroquine resistance transporter
Supporting Information Available
Chemistry, culturing of parasites, and synchrotron X-ray fluorescence (XRF) studies. This material is available free of charge via the Internet at http://pubs.acs.org.
This study was funded by a grant from the Ministère de l'Enseignement Supérieur to F.D.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. World Malaria Report 2011; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Mita T.; Tanabe K.; Kita K. Spread and Evolution of Plasmodium falciparum Drug Resistance. Parasitol. Int. 2009, 58, 201–209. [DOI] [PubMed] [Google Scholar]
- Yuan J.; Cheng K. C.-C.; Johnson R. L.; Huang R.; Pattaradilokrat S.; Liu A.; Guha R.; Fidock D. A.; Inglese J.; Wellems T. E.; et al. Chemical Genomic Profiling for Antimalarial Therapies, Response Signatures, and Molecular Targets. Science 2011, 333, 724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombo-Ngoma G.; Supan C.; Dal-Bianco M. P.; Missinou M. A.; Matsiegui P.-B.; Ospina Salazar C. L.; Issifou S.; Ter-Minassian D.; Ramharter M.; Kombila M.; et al. Phase I Randomized Dose-ascending Placebo-controlled Trials of Ferroquine—a Candidate Anti-malarial Drug—in Adults with Asymptomatic Plasmodium falciparum Infection. Malar. J. 2011, 10, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubar F.; Bohic S.; Slomianny C.; Morin J.-C.; Thomas P.; Kalamou H.; Guérardel Y.; Cloetens P.; Khalife J.; Biot C. In Situ Nanochemical Imaging of Label-free Drugs: a Case Study of Antimalarials in Plasmodium falciparum-infected Erythrocytes. Chem. Commun. (Cambridge, U. K.) 2012, 48, 910–912. [DOI] [PubMed] [Google Scholar]
- Sullivan D. J.; Gluzman I. Y.; Russell D. G.; Goldberg D. E. On the Molecular Mechanism of Chloroquine's Antimalarial Action. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 11865–11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepe P. D. PfCRT-Mediated Drug Transport in Malarial Parasites. Biochemistry 2011, 50, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary T. G.; Jensen J. B.; Ginsburg H. Uptake of [3H]chloroquine by Drug-sensitive and -resistant Strains of the Human Malaria Parasite Plasmodium falciparum. Biochem. Pharmacol. 1986, 35, 3805–3812. [DOI] [PubMed] [Google Scholar]
- Krogstad D. J.; Gluzman I. Y.; Kyle D. E.; Oduola A. M.; Martin S. K.; Milhous W. K.; Schlesinger P. H. Efflux of Chloroquine from Plasmodium falciparum: Mechanism of Chloroquine Resistance. Science 1987, 238, 1283–1285. [DOI] [PubMed] [Google Scholar]
- Bray P. G.; Howells R. E.; Ritchie G. Y.; Ward S. A. Rapid Chloroquine Efflux Phenotype in Both Chloroquine-sensitive and Chloroquine-resistant Plasmodium falciparum. A Correlation of Chloroquine Sensitivity with Energy-dependent Drug Accumulation. Biochem. Pharmacol. 1992, 44, 1317–1324. [DOI] [PubMed] [Google Scholar]
- Paguio M. F.; Cabrera M.; Roepe P. D. Chloroquine Transport in Plasmodium falciparum. 2. Analysis of PfCRT-mediated Drug Transport Using Proteoliposomes and a Fluorescent Chloroquine Probe. Biochemistry 2009, 48, 9482–9491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A. B. S.; Verdier-Pinard D.; Fidock D. A. Chloroquine Resistance in Plasmodium falciparum Malaria Parasites Conferred by Pfcrt Mutations. Science 2002, 298, 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C. P.; Stein W. D.; Lanzer M. Is PfCRT a Channel or a Carrier? Two Competing Models Explaining Chloroquine Resistance in Plasmodium falciparum. Trends Parasitol. 2007, 23, 332–339. [DOI] [PubMed] [Google Scholar]
- Martin R. E.; Marchetti R. V.; Cowan A. I.; Howitt S. M.; Bröer S.; Kirk K. Chloroquine Transport via the Malaria Parasite’s Chloroquine Resistance Transporter. Science 2009, 325, 1680–1682. [DOI] [PubMed] [Google Scholar]
- Fidock D. A.; Nomura T.; Talley A. K.; Cooper R. A.; Dzekunov S. M.; Ferdig M. T.; Ursos L. M.; Sidhu A. B.; Naudé B.; Deitsch K. W.; et al. Mutations in the P. Falciparum Digestive Vacuole Transmembrane Protein PfCRT and Evidence for Their Role in Chloroquine Resistance. Mol. Cell 2000, 6, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper R. A.; Ferdig M. T.; Su X.-Z.; Ursos L. M. B.; Mu J.; Nomura T.; Fujioka H.; Fidock D. A.; Roepe P. D.; Wellems T. E. Alternative Mutations at Position 76 of the Vacuolar Transmembrane Protein PfCRT Are Associated with Chloroquine Resistance and Unique Stereospecific Quinine and Quinidine Responses in Plasmodium falciparum. Mol. Pharmacol. 2002, 61, 35–42. [DOI] [PubMed] [Google Scholar]
- Baro N. K.; Pooput C.; Roepe P. D. Analysis of Chloroquine Resistance Transporter (CRT) Isoforms and Orthologues in S. Cerevisiae Yeast. Biochemistry 2011, 50, 6701–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Paguio M.; Roepe P. D. The Antimalarial Drug Resistance Protein Plasmodium falciparum Chloroquine Resistance Transporter Binds Chloroquine. Biochemistry 2004, 43, 8290–8296. [DOI] [PubMed] [Google Scholar]
- Lekostaj J. K.; Natarajan J. K.; Paguio M. F.; Wolf C.; Roepe P. D. Photoaffinity Labeling of the Plasmodium falciparum Chloroquine Resistance Transporter with a Novel Perfluorophenylazido Chloroquine. Biochemistry 2008, 47, 10394–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubar F.; Egan T. J.; Pradines B.; Kuter D.; Ncokazi K. K.; Forge D.; Paul J.-F.; Pierrot C.; Kalamou H.; Khalife J.; et al. The Antimalarial Ferroquine: Role of the Metal and Intramolecular Hydrogen Bond in Activity and Resistance. ACS Chem. Biol. 2011, 6, 275–287. [DOI] [PubMed] [Google Scholar]
- Casabianca L. B.; An D.; Natarajan J. K.; Alumasa J. N.; Roepe P. D.; Wolf C.; Dios A. C. de Quinine and Chloroquine Differentially Perturb Heme Monomer-dimer Equilibrium. Inorg. Chem. 2008, 47, 6077–6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohic S.; Cotte M.; Salomé M.; Fayard B.; Kuehbacher M.; Cloetens P.; Martinez-Criado G.; Tucoulou R.; Susini J. Biomedical Applications of the ESRF Synchrotron-based Microspectroscopy Platform. J. Struct. Biol. 2012, 177, 248–258. [DOI] [PubMed] [Google Scholar]
- Henry M.; Briolant S.; Fontaine A.; Mosnier J.; Baret E.; Amalvict R.; Fusaı T.; Fraisse L.; Rogier C.; Pradines B. In Vitro Activity of Ferroquine Is Independent of Polymorphisms in Transport Protein Genes Implicated in Quinoline Resistance in Plasmodium falciparum. Antimicrob. Agents Chemother. 2008, 52, 2755–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg H.; Famin O.; Zhang J.; Krugliak M. Inhibition of Glutathione-dependent Degradation of Heme by Chloroquine and Amodiaquine as a Possible Basis for Their Antimalarial Mode of Action. Biochem. Pharmacol. 1998, 56, 1305–1313. [DOI] [PubMed] [Google Scholar]
- Meister A.; Anderson M. E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [DOI] [PubMed] [Google Scholar]
- Kehr S.; Sturm N.; Rahlfs S.; Przyborski J. M.; Becker K. Compartmentation of Redox Metabolism in Malaria Parasites. PLoS Pathog. 2010, 6, e1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzewitz E.-M.; Wong E. H.; Müller S. Dissecting the Role of Glutathione Biosynthesis in Plasmodium falciparum. Mol. Microbiol. 2012, 83, 304–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers R. L.; Nash M. N.; Martin R. E.. Know Your Enemy: Understanding the Role of PfCRT in Drug Resistance Could Lead to New Antimalarial Tactics. Cell. Mol. Life Sci. 2012. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.