

Abstract
There is a desperate need to develop new antibiotic agents to combat the rise of drug-resistant bacteria, such as clinically important Staphylococcus aureus. The essential multifunctional enzyme, biotin protein ligase (BPL), is one potential drug target for new antibiotics. We report the synthesis and characterization of a series of biotin analogues with activity against BPLs from S. aureus, Escherichia coli, and Homo sapiens. Two potent inhibitors with Ki < 100 nM were identified with antibacterial activity against a panel of clinical isolates of S. aureus (MIC 2–16 μg/mL). Compounds with high ligand efficiency and >20-fold selectivity between the isozymes were identified and characterized. The antibacterial mode of action was shown to be via inhibition of BPL. The bimolecular interactions between the BPL and the inhibitors were defined by surface plasmon resonance studies and X-ray crystallography. These findings pave the way for second-generation inhibitors and antibiotics with greater potency and selectivity.
Keywords: biotin protein ligase, enzyme, enzyme inhibitor, antibiotic, medicinal chemistry
It is imperative that we identify new antibiotics to combat drug-resistant bacteria.1 One clinically important pathogen, Staphylococcus aureus (S. aureus), is responsible for more than half of all life-threatening bloodstream infections. S. aureus is challenging to control as it has the ability to rapidly acquire drug-resistance mechanisms in both hospitals and the community, making treatment increasingly difficult and costly.2,3 An important strategy to combat drug resistance is to develop novel antibiotic classes for which there are no pre-existing resistance mechanisms. This is becoming increasingly difficult, as most of the known chemical classes and obvious drug targets have been well explored, leaving the more challenging targets as a new frontier for antibacterial research. For example, little work has been done on essential bacterial enzymes that have mammalian paralogues due to perceived fears of possible toxicity.4 For these targets, selective inhibition is critical.
The ubiquitous enzyme biotin protein ligase (BPL) is one potential antibacterial target that has not yet been comprehensively investigated.5 BPL is responsible for the attachment of the cofactor biotin onto biotin-dependent enzymes. This proceeds in two partial reactions. Initially, the adenylated reaction intermediate biotinyl-5′-AMP is produced from biotin and ATP. Subsequently, the biotin moiety is covalently attached to the ε-amino group of a single target lysine residue present in the active site of biotin-dependent enzymes.6 Here, biotin is required to facilitate the carboxylation of metabolites. Without the attached cofactor, biotin-dependent enzymes are catalytically inactive and unable to fulfill their critical metabolic roles. As all organisms possess between one and five biotin-dependent enzymes, all organisms require a BPL, as there are no alternative enzymes to perform protein biotinylation. One important biotin-dependent enzyme is acetyl CoA carboxylase, which catalyzes the first committed step in the fatty acid biosynthetic pathway.4 As this pathway is essential for bacterial cell membrane maintenance and biogenesis, they are a potential source of new antibiotic targets in certain Gram-positive bacteria such as S. aureus.7S. aureus expresses a second biotin-dependent enzyme, pyruvate carboxylase, which catalyzes the conversion of pyruvate to oxaloacetate, thereby replenishing the TCA cycle.8 In addition to its biotin ligase activity, certain bacterial BPLs also function as transcriptional repressors, making them bifunctional proteins.9 BPL recognition sites in the S. aureus genome suggest that S. aureus BPL regulates expression of the enzymes in the biotin biosynthesis operon, as well as the biotin transport protein BioY.10 Thus, BPL is the master regulator protein of all biotin-mediated events in bacteria and, therefore, an attractive new antibiotic drug target.
In this paper, we present a novel series of biotin analogues with potent inhibitory activity against SaBPL and antibacterial activity against clinical strains of S. aureus. The mode of small molecule binding to SaBPL was defined by X-ray crystallography and surface plasmon resonance studies. A few examples of biotin analogues with antibacterial activity do appear in the literature, including the natural product α-dehydrobiotin11 and an analogue chemically modified at the N1 position required for binding carbon dioxide (Figure 1).12 These function by first being incorporated into a biotin-dependent enzyme where they abolish catalytic activity by preventing the binding or transfer of carbon dioxide.13−15 Importantly, these analogues are either not specific or loose broad-spectrum antibacterial activity when assayed in rich growth media containing biotin.11,14 Moreover, their activity has not been linked to the inhibition of BPL as they only transiently occupy the active site of this enzyme. A more effective approach for antibiotic discovery requires high-affinity inhibitors that bind directly and specifically to the bacterial BPL target, thereby preventing all protein biotinylation. A proof of concept for this approach to antibiotic discovery has been reported recently, targeting the BPLs from Mycobacterium tuberculosis and S. aureus.5,16 In this current paper, we investigated biotin analogues with a view to identifying BPL inhibitors with improved ligand efficiency (LE). Our findings provide new lead structures for further optimization, for example, 5 and 16.
Figure 1.

Chemical structures of (a) biotin 1 and its analogues (b) α-dehydrobiotin, (c) N1 substituted biotin, and (d) the pharmacophore investigated in this study.
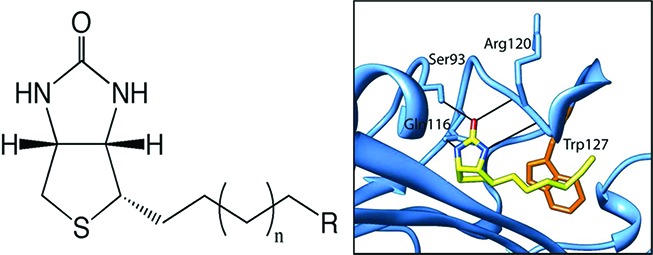
We began our search by screening analogues of biotin for their inhibitory properties. The compounds were assayed in an in vitro biotinylation assay measuring the incorporation of radiolabeled biotin into protein.17 Purified recombinant BPLs from the Gram-positive bacterium S. aureus (SaBPL18), the Gram-negative bacterium Escherichia coli (EcBPL19), and Homo sapiens (HsBPL20) were all assessed to investigate potency and species selectivity (Table 1). Ki values were calculated assuming that the mechanism of action was competitive with biotin, in agreement with Lineweaver–Burk analysis (Supporting Information, Figure S1).21 This identified biotinol (6) as a pan inhibitor with Ki values of 3.4–4 μM for all three BPLs (Table 1). The binding mechanism was confirmed by solving the X-ray crystal structure of SaBPL in complex with 6 (Figure 2a). Consistent with other BPL crystal structures that appear in the protein database, our data revealed the biotin-binding pocket of SaBPL to be relatively small (187 Å3) with a hydrophobic cavity encasing the polar ureido and thiophene rings of biotin. This suggested limited opportunity for chemical modification of the biotin heterocycle in our inhibitor design. A narrow hydrophobic tunnel accommodates the valeric acid side chain with the carboxyl group being solvent exposed to provide a prime target for derivitisation. The protein:ligand interaction is stabilized through hydrogen bonding between the hydroxyl group (present on biotin and alcohol 6) and an NH backbone amide of Arg 122 (Figure 2a). A series of analogues were proposed to further explore inhibition of BPL as a route to novel antibacterial agents. Our hypothesis was that derivatization of the carboxyl moiety of biotin would produce tight-binding inhibitors with increased potency and selectivity. Thus, to establish optimal binding, modifications of biotin were investigated that either replaced the carboxyl functionality with motifs capable of forming direct contacts with BPL as in compounds 3, 16, and 5, and/or modified the length of the carbon chain (compounds 7, 11, 17, 14, and 15).
Table 1. SAR Data for the Biotin Analogue Seriesa.
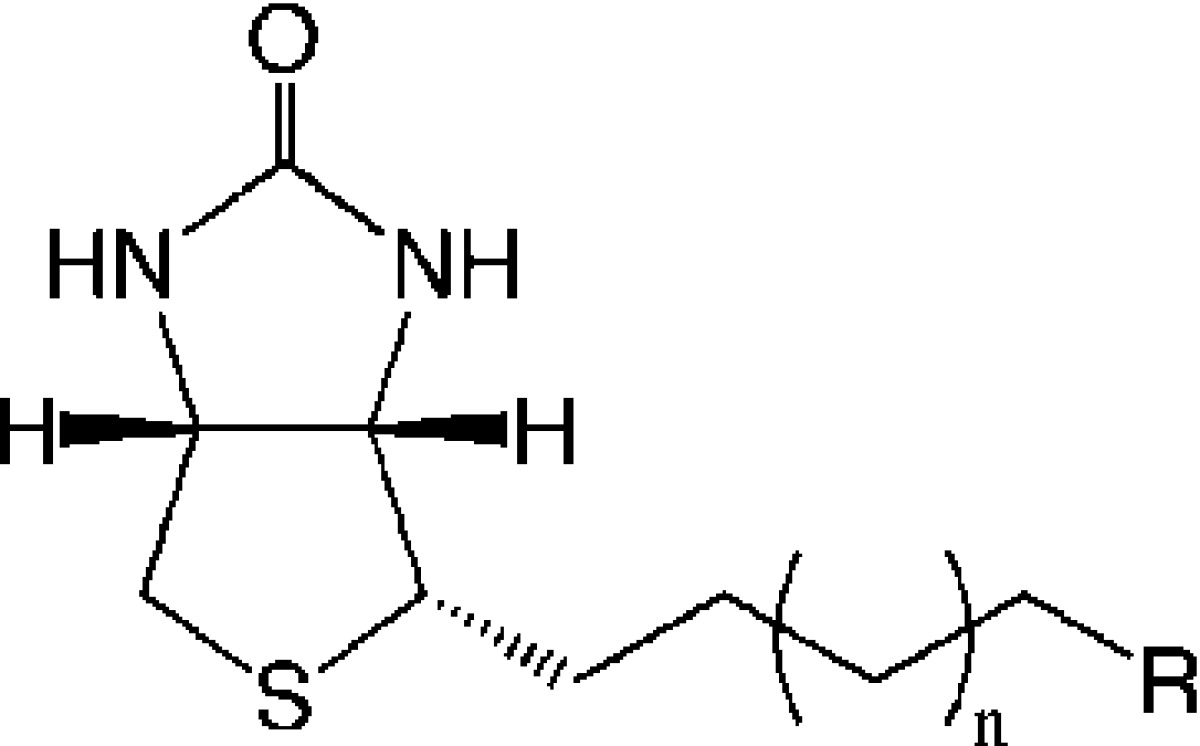
|
S. aureus |
E. coli |
H. sapiens | selectivityc | |||||
|---|---|---|---|---|---|---|---|---|
| ID | n | R | Ki (μM) | LEb | Ki (μM) | LEb | Ki (μM) | Hs vs Sa/Ec vs Hs/Ec vs Sa |
| 6 | 2 | OH | 3.37 ± 0.3 | 0.52 | 3.96 ± 0.4 | 0.51 | 3.85 ± 0.3 | 1.1/1.0/1.2 |
| 7 | 3 | OH | >20 | ND | >20 | ND | >20 | ND |
| 3 | 1 | NH2 | >20 | ND | >20 | ND | >20 | ND |
| 11 | 2 | NH2 | >20 | ND | >20 | ND | >20 | ND |
| 16 | 1 | CH3 | 0.05 ± 0.01 | 0.69 | 1.10 ± 0.12 | 0.56 | 0.14 ± 0.02 | 2.8/7.9/22 |
| 17 | 2 | CH3 | 0.52 ± 0.06 | 0.56 | 7.30 ± 1.9 | 0.46 | 6.43 ± 1.4 | 12.4/1.1/14 |
| 5 | 1 | C≡CH | 0.08 ± 0.01 | 0.67 | 0.90 ± 0.1 | 0.57 | 0.2 ± 0.03 | 2.5/4.5/11.3 |
| 14 | 2 | C≡CH | 0.3 ± 0.05 | 0.58 | 7.33 ± 1.0 | 0.46 | 3.50 ± 0.5 | 11.7/2.1/24.4 |
| 15 | 3 | C≡CH | 2.40 ± 0.02 | 0.47 | 20.0 ± 0.3 | 0.39 | 12.0 ± 1.83 | 5/1.7/8.3 |
A SAR series was investigated based upon the pharmacophore shown above. Compounds were assayed against BPLs from three species and where inhibition was observed Ki values are reported. Each Ki value is the mean (±standard error) of three independent experiments.
LE is ligand efficiency in kcal K–1 mol–1 per heavy atom.
Selectivity is expressed as ratios of Ki values. Sa is S. aureus BPL, Ec is E. coli BPL, and Hs is H. sapiens BPL.
Figure 2.
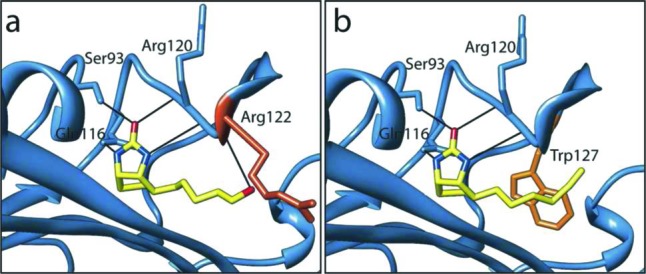
X-ray structures of inhibitors in complex with BPL. S. aureus BPL was crystallized in complex with biotin analogues alcohol 6 (a) and acetylene 14 (b). The hydrogen-bonding interactions with the inhibitors are shown.
The syntheses of all of the derivatives are shown in Scheme 1. Biotin 1 and homobiotin 2(5) were first esterified, and the resulting methyl esters reduced with LiAlH4 to give the alcohols 6 and 7, respectively. Tosylation followed by bromination then gave 12 and 13, respectively, which were separately reacted with lithium-acetylide EDA complex to give the two acetylenes 14 and 15.5 The tosylate 8 was also reacted with NaN3 to give the azide 10, which then gave the amine 11 using Staudinger reduction with PPh3 followed by hydrolysis of the aza-ylide intermediate. The two tosylates 8 and 9 were also reduced with LiAlH4 to give the alkyl derivatives 16 and 17, respectively. Curtius rearrangement of biotin 1, on reaction with diphenylphosphoryl azide in tert-butanol, followed by treatment with 6 N aqueous HCl, gave amine 3.22 Finally, bromide 4 was prepared by Barton decarboxylation of 1 using 2-pyrithione, which on treatment with lithium-acetylide EDA complex gave acetylene 5.5
Scheme 1.

The biotin derivatives were tested against the three BPLs using an in vitro biotinylation assay (Table 1) to reveal potent and selective inhibitors of SaBPL. Interestingly, increasing the chain length of 6 by one carbon resulted in an inactive compound (7). The crystallographic data for SaBPL bound to 6 as discussed earlier supports this observation, where the key hydrogen bond between the hydroxyl group of 6 and the enzyme is incompatible with the longer chain length. Replacement of the hydroxyl group of 6 with an amine, as in 3 and 11, removed all inhibitory activity. This was somewhat surprising given that the amine would be expected to have similar polarity and hydrogen-bonding properties to the hydroxyl group of 6. This observation may simply reflect the relative pKa values of the two groups. We also assayed the synthetic intermediate bromides (4, 12, and 13), and these were all devoid of activity, presumably due to the steric bulk of the halids.
We next investigated hydrophobic groups (alkyl and acetylene) at the terminus of the carbon chain of biotin (see 5 and 14–17) that we anticipated might be accommodated by the hydrophobic nature of the biotin pocket. Most notably 5, 14, 16, and 17 exhibited up to a 20-fold selectivity for SaBPL over EcBPL and HsBPL. Interestingly, removal of the tetrahydrothiophane ring in 14 removed all activity (see 18 and 19), highlighting the importance of the fused bicyclic ring of biotin. In the alkyl series, 16 was the most potent inhibitor for all three enzymes (SaBPL, Ki = 0.05 μM) and displayed the greatest LE (0.69 kcal K–1 mol–1 per heavy atom). Again, a longer chain length (see 17) decreased potency by ≈10-fold for the two bacterial enzymes and by 45-fold for the human enzyme. However, significant improvement in selectivity was apparent (see Table 1). The most potent compound in the acetylene series was again that with the shortest carbon chain (see 5, Ki = 0.08 μM and LE 0.67 kcal K–1 mol–1 per heavy atom for SaBPL) with an observed decrease in potency and LE on increasing the chain length from n = 1 to 3 (see compounds 14 and 15 in Table 1). Once again, the most selective compound was where n = 2 (see 14; with 20- and 10-fold selectivity for SaBPL against E. coli and H. sapiens BPL, respectively). The acetylene 5 retained significant potency against the target SaBPL with a Ki = 0.30 μM. The structure of biotin acetylene (14) in complex with SaBPL was determined to confirm both the mode of binding and our proposal that substitution of the carboxyl group of biotin with hydrophobic functionalities would promote binding to the target. In agreement with the hypothesis, the complex was stabilized through a direct hydrophobic interaction between the acetylene and the Trp 127 (Figure 2b). Given that we observed selectivity in this series, we propose that this interaction should be further explored in inhibitor design. Our data strongly suggest that the biotin pockets of the three BPLs are not as structurally conserved as previously proposed, and this could be explored to improve potency and selectivity.
Ideally, our antibacterial compounds should have a relatively high affinity for BPL through slow dissociation from the binding pocket. To study this, SaBPL was immobilized on a sensor chip for subsequent binding studies by surface plasmon resonance. Biotin and analogues 6, 16, and 5 were then separately passed across the surface, providing quantitative analysis of association and dissociation rates. The binding of biotin to SaBPL (Figure 3a) was found to exhibit rapid on and off rates outside the range for quantitative evaluation. Hence, the KD (9.2 ± 1.4 μM) was calculated using steady-state affinity. This observation is consistent with a two-step binding mechanism that has been reported for E. coli BPL.23 Initially, the enzyme and biotin rapidly form a “collision complex” that is followed by a slower conformational change to BPL that is both rate-limiting in the binding mechanism and necessary to stablize the enzyme–biotin complex. Alcohol analogue 6 showed similar kinetics of binding as biotin (Figure 3b, KD = 2.5 ± 0.3 μM), representative of a dynamic equilibrium between the free enzyme and the collision complex but unable to overcome the rate-limiting conformational change. In contrast, 16 and 5 displayed slower association (ka = 43.2 ± 2.3 × 103 M–1 s–1 and 41.2 ± 2.3 × 103 M–1 s–1, respectively) and dissociation rates relative to both biotin and 6 (16kd = 2.80 ± 0.20 × 10–3 s–1 and 5 3.40 ± 0.30 × 10–3 s–1), thereby contributing to their significantly higher affinities (16KD = 0.06 ± 0.01 μM, Figure 3c, and 5KD = 0.08 ± 0.01, Figure 3d). The different association and dissociation kinetics displayed by 16 and 5 as compared to biotin and 6 imply that the modes of binding are mechanistically different. We propose that unlike biotin, 16 and 5 do indeed induce the conformational changes to SaBPL that are necessary to retard the inhibitors from vacating their binding pockets. For antibacterial discovery, inhibitors that possess high occupancy rates on their bacterial targets are desirable.
Figure 3.
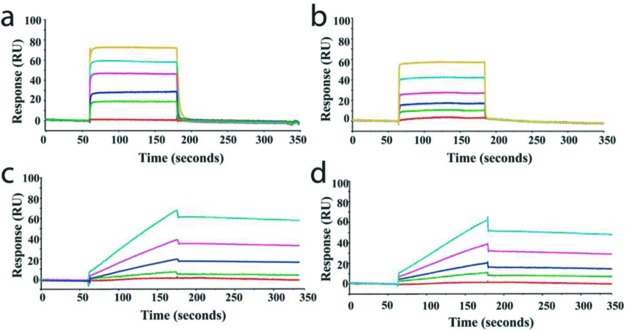
SPR analysis of BPL inhibitor binding. Surface plasmon resonance sensorgrams are shown for the binding of (a) biotin, (b) 6, (c) 16, and (d) 5 to immobilized S. aureus BPL. Varying concentrations of the inhibitors were included in the running buffer, and analysis was performed as described in the Supporting Information.
Finally, the antibacterial activity of the most potent biotin analogues was determined using antimicrobial growth assays. These studies revealed significant bacteriostatic activity against both Gram-positive and Gram-negative bacteria. Compounds 16 and 5 were the most potent against clinical isolates of S. aureus with MICs in the range of 2–16 μg/mL, including methicillin-susceptible and -resistant strains (Table 2). This is consistent with the fact that these two derivatives were the most potent inhibitors of SaBPL (Table 1). Derivative 17, containing an extra carbon in the side chain relative to 16, was less active against MSSA and MRSA, but interestingly, it was weakly active against vancomycin-resistant Enterococcus (MIC 32 μg/mL). Compounds 16 and 5 were also active against coagulase negative Staphylococci strains. The mechanism of antibacterial action was assessed with a complementation assay using 5 with an E. coli K12 strain engineered to overexpress the BPL target. Bacterial growth was monitored over 14 h in the absence or presence of 5 at 32 μg/mL (Figure S2 in the Supporting Information). Overexpression of EcBPL completely alleviated antibacterial activity, strongly implying that the drug target is indeed BPL. Finally, the toxicity of the compounds was addressed in a mammalian cell culture model using HepG2 cells. These studies showed that the metabolic activity of the cells was uneffected when treated with 64 μg/mL of 7, 16, 17, 5, 14, and 15.
Table 2. Antibacterial Susceptibility Testinga.
| MIC50 (μg/mL) |
MIC90 (μg/mL) |
||||
|---|---|---|---|---|---|
| strain | range (μg/mL) | 16 | 5 | 16 | 5 |
| MSSA (n = 8) | 4–16 | 8 | 8 | 16 | 16 |
| MRSA (n = 8) | 4–16 | 8 | 8 | 16 | 16 |
| coagulase negative Staphylococci (n = 7) |
2–>64 | 2 | 4 | 8 | 8 |
MICs are shown for a library of S. aureus clinical isolates of coagulase negative and positive strains, including methicillin-sensitive (MSSA) and -resistant (MRSA) subtypes.
Here, we report new data that supports the hypothesis that BPL is a druggable antibacterial target in vitro. Our data demonstrate that BPL inhibitors with favorable in vitro properties also show significant antibacterial activity against clinical isolates of methicillin-sensitive and -resistant S. aureus. It is noteworthy that there was a positive correlation between slow enzyme:inhibitor dissociation kinetics and potent antibacterial activity. The quantitative data reported here help to define the target product profile necessary for future chemical optimization. Our biotin alkyl and acetylene series represent new chemical scaffolds with high LE for chemical development toward new antibacterial agents, a point that we have begun to address as reported in our earlier publication.5
Acknowledgments
Diffraction data were collected on the MX1 beamline at the Australian Synchrotron, Victoria, Australia.
Glossary
Abbreviations
- BPL
biotin protein ligase
- Ec
Escherichia coli
- Hs
Homo sapiens
- LE
ligand efficiency
- MIC
minimal inhibitory concentration
- Sa
Staphylococcus aureus
Supporting Information Available
Assay and synthetic procedures and data for selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
We acknowledge funding from the Australian Research Council and National Health and Medical Research Council of Australia (application number 1011806), BioInnovationSA, and the University of Adelaide's Commercial Accelerator Scheme.
The authors declare no competing financial interest.
Author Status
M.C.J.W. is a National Health and Medical Research Council Senior Research Fellow.
Author Present Address
⊥ School of Materials Science and Engineering, Nanyang Technological University, Singapore. E-mail: OJZVAREC@ntu.edu.sg.
Author Contributions
# These authors contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supplementary Material
References
- Cooper M. A.; Shlaes D. Fix the antibiotics pipeline. Nature 2011, 472734132. [DOI] [PubMed] [Google Scholar]
- Allegranzi B.; Bagheri Nejad S.; Combescure C.; Graafmans W.; Attar H.; Donaldson L.; Pittet D. Burden of endemic health-care-associated infection in developing countries: Systematic review and meta-analysis. Lancet 2011, 3779761228–241. [DOI] [PubMed] [Google Scholar]
- Jean S. S.; Hsueh P. R. High burden of antimicrobial resistance in Asia. Int. J. Antimicrob. Agents 2011, 374291–295. [DOI] [PubMed] [Google Scholar]
- Polyak S. W.; Abell A. D.; Wilce M. C.; Zhang L.; Booker G. W. Structure, function and selective inhibition of bacterial acetyl-CoA carboxylase. Appl. Microbiol. Biotechnol. 2012, 933983–992. [DOI] [PubMed] [Google Scholar]
- Soares da Costa T. P.; Tieu W.; Yap M. Y.; Pendini N. R.; Polyak S. W.; Sejer Pedersen D.; Morona R.; Turnidge J. D.; Wallace J. C.; Wilce M. C.; Booker G. W.; Abell A. D. Selective inhibition of biotin protein ligase from Staphylococcus aureus. J. Biol. Chem. 2012, 287, 17823–17832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendini N. R.; Bailey L. M.; Booker G. W.; Wilce M. C.; Wallace J. C.; Polyak S. W. Microbial biotin protein ligases aid in understanding holocarboxylase synthetase deficiency. Biochim. Biophys. Acta 2008, 17847–8973–982. [DOI] [PubMed] [Google Scholar]
- Parsons J. B.; Rock C. O. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery?. Curr. Opin. Microbiol. 2011, 145544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jitrapakdee S.; St. Maurice M.; Rayment I.; Cleland W. W.; Wallace J. C.; Attwood P. V. Structure, mechanism and regulation of pyruvate carboxylase. Biochem. J. 2008, 4133369–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett D. Biotin sensing at the molecular level. J. Nutr. 2009, 1391167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov D. A.; Mironov A. A.; Gelfand M. S. Conservation of the biotin regulon and the BirA regulatory signal in Eubacteria and Archaea. Genome Res. 2002, 12101507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanka L. J.; Bergy M. E.; Kelly R. B. Naturally occurring antimetabolite antibiotic related to biotin. Science 1966, 1547571667–1668. [DOI] [PubMed] [Google Scholar]
- Amspacher D. R.; Blanchard C. Z.; Fronczek F. R.; Saraiva M. C.; Waldrop G. L.; Strongin R. M. Synthesis of a reaction intermediate analogue of biotin-dependent carboxylases via a selective derivatization of biotin. Org. Lett. 1999, 1199–102. [DOI] [PubMed] [Google Scholar]
- Blanchard C. Z.; Amspacher D.; Strongin R.; Waldrop G. L. Inhibition of biotin carboxylase by a reaction intermediate analog: Implications for the kinetic mechanism. Biochem. Biophys. Res. Commun. 1999, 2662466–471. [DOI] [PubMed] [Google Scholar]
- Levert K. L.; Waldrop G. L.; Stephens J. M. A biotin analog inhibits acetyl-CoA carboxylase activity and adipogenesis. J. Biol. Chem. 2002, 2771916347–16350. [DOI] [PubMed] [Google Scholar]
- Piffeteau A.; Dufour M. N.; Zamboni M.; Gaudry M.; Marquet A. Mechanism of the antibiotic action of alpha-dehydrobiotin. Biochemistry 1980, 19133069–3073. [DOI] [PubMed] [Google Scholar]
- Duckworth B. P.; Geders T. W.; Tiwari D.; Boshoff H. I.; Sibbald P. A.; Barry C. E. 3rd; Schnappinger D.; Finzel B. C.; Aldrich C. C. Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis. Chem. Biol. 2011, 18111432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak S. W.; Chapman-Smith A.; Brautigan P. J.; Wallace J. C. Biotin protein ligase from Saccharomyces cerevisiae. The N-terminal domain is required for complete activity. J. Biol. Chem. 1999, 2744632847–32854. [DOI] [PubMed] [Google Scholar]
- Pendini N. R.; Polyak S. W.; Booker G. W.; Wallace J. C.; Wilce M. C. Purification, crystallization and preliminary crystallographic analysis of biotin protein ligase from Staphylococcus aureus. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2008, 64Part 6520–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman-Smith A.; Mulhern T. D.; Whelan F.; Cronan J. E. Jr.; Wallace J. C. The C-terminal domain of biotin protein ligase from E. coli is required for catalytic activity. Protein Sci. 2001, 10122608–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayende L.; Swift R. D.; Bailey L. M.; Soares da Costa T. P.; Wallace J. C.; Booker G. W.; Polyak S. W. A novel molecular mechanism to explain biotin-unresponsive holocarboxylase synthetase deficiency. J. Mol. Med. (Berlin) 2012, 90181–88. [DOI] [PubMed] [Google Scholar]
- Cleland W. W. Steady State Kinetics. The Enzymes 1970, 2, 1. [Google Scholar]
- Szalecki W. Synthesis of norbiotinamine and its derivatives. Bioconjugate Chem. 1996, 72271–273. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Nenortas E.; Beckett D. Evidence for distinct ligand-bound conformational states of the multifunctional Escherichia coli repressor of biotin biosynthesis. Biochemistry 1995, 345116624–16631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.