

Abstract

The ERK/MAP kinase cascade is a key mechanism subject to dysregulation in cancer and is constitutively activated or highly upregulated in many tumor types. Mutations associated with upstream pathway components RAS and Raf occur frequently and contribute to the oncogenic phenotype through activation of MEK and then ERK. Inhibitors of MEK have been shown to effectively block upregulated ERK/MAPK signaling in a range of cancer cell lines and have further demonstrated early evidence of efficacy in the clinic for the treatment of cancer. Guided by structural insight, a strategy aimed at the identification of an optimal diphenylamine-based MEK inhibitor with an improved metabolism and safety profile versus PD-0325901 led to the discovery of development candidate 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2-yl]azetidin-3-ol (XL518, GDC-0973) (1). XL518 exhibits robust in vitro and in vivo potency and efficacy in preclinical models with sustained duration of action and is currently in early stage clinical trials.
Keywords: MAPK pathway, MEK, cancer, kinase inhibitor, XL518, GDC-0973
The MAPK cascade, or mitogen-activated protein kinase signal transduction pathway, is a mechanism commonly subject to dysregulation in cancer, and constitutive or highly upregulated signaling is a frequent hallmark of oncogenic transformation and progression. Controlled by activation of RAS at the cell surface interior, the subsequent stimulation of Raf and then MEK and ERK serves to regulate a range of key intracellular effectors associated with cell proliferation,1 invasion,2 angiogenesis,3 and apoptotic resistance.4 Mutated RAS is associated with almost one-third of human cancers, and a majority of malignant melanomas and papillary thyroid cancers harbor B-Raf mutations.5,6 Inhibitors of MEK have demonstrated efficacy against malignant tumors characterized by mutations in either RAS or Raf in preclinical models, and early development candidates including GSK1120212,7 AZD6244,8 and PD0325901 (2)9 have further demonstrated evidence of activity in the clinical setting as well.
In some tumors, activation of both the RAS driven ERK/MAPK cascade and the PI3K-Akt pathway is observed, resulting in degenerate and convergent oncogenic signals, and upregulation or constitutive PI3K pathway activation is associated with resistance to MEK inhibitor single agent treatment.10 Several combination approaches using inhibitors of mTor, PI3K, Akt, and Raf have more recently been validated in preclinical models and are being pursued clinically. Herein, the discovery of XL518 (GDC-0973) (1), a potent and selective MEK inhibitor, is described. XL518 is currently in early stage clinical testing as both a single agent and in combination with the class I PI3K inhibitor GDC-0941.11
Our goal at the outset was the identification of a potent
and selective
MEK inhibitor with sustained duration of efficacy suitable for qd
dosing and an optimized safety profile relative to clinical precursors.
The diphenylamine series disclosed by the Pfizer/Warner Lambert groups
served as a starting point for our effort. A key aspect that served
as a guide post for the optimization process included elimination
of the hydroxamate ester, which demonstrated metabolic instability
in the CI-1040 series.12 Potentially neurological
side effects associated with ataxia, confusion, and syncope were also
observed in patients during early trials involving PD-0325901 (2), possibly associated with BBB penetration and MAPK pathway
suppression in brain tissue by parent or active metabolite (4), and we sought to minimize this activity as well.13
Guided by structural insight, our approach focused on replacement of the hydroxamate ester with a carboxamide. Binding of the hydroxamate group in 2 is defined by a network of hydrogen bond contacts involving the C2 and C3 hydroxyl groups with the γ-phosphate of bound ATP (Figure 1). The diphenylamine core binds in the manner reported for early analogues in the CI-1040 series.14 Initial efforts focused on the synthesis and evaluation of simple cyclic and acyclic carboxamide derivatives of (4) that feature polar substituents including alcohols (Scheme 1). The set of amide derivatives (6–19) featured in Table 1 were evaluated for MEK biochemical inhibitory activity in a c-Raf-1/MEK1/ERK triple coupled assay using an AlphaScreen chemiluminescense readout and for cellular activity in MDA-MB-231T breast adenocarcinoma cells, selected for the expression of both K-RAS G13D and B-RAF G464V mutations.
Figure 1.
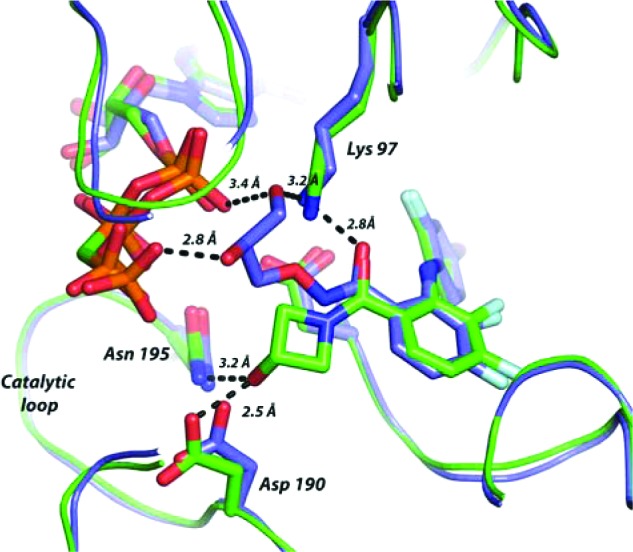
Overlay of MEK1:AMP-PCP complex X-ray cocrystal structures for PD0325901 (2) in blue and azetidinol (7) in green. Dashed lines indicate key contacts and distances for the hydroxamate and carboxamide.
Scheme 1. General Synthesis of Carboxamide MEK Inhibitors.

Reagents and conditions: (a) Compound 1, cyanuric fluoride, DCM, pyr. 0 °C. (b) Compound 5, DMF, DIPEA, RT. (c) Compound 4, PyBOP, DMF, DIPEA, RT.
Table 1. In Vitro Activity for MEK Inhibitors.
| compd | MEK1 IC50 (nM)a | MD-MBA-231T IC50 (nM)b |
|---|---|---|
| 1 | 0.9 | 0.2 |
| 2 | 0.6 | 0.9 |
| 3 | 801 | 128 |
| 4 | 19.5 | 102 |
| 6 | 535 | 111 |
| 7 | 44 | 29 |
| 8 | 6.6 | 57 |
| 9 | 82 | 51 |
| 10 | 27 | 29 |
| 11 | 220 | 81 |
| 12 | 7.2 | 6.9 |
| 13 | 1.8 | 3.7 |
| 14 | 4.8 | 11 |
| 15 | 5.1 | 14 |
| 16 | 7.6 | 50 |
| 17 | 1708 | 331 |
| 18 | 5.2 | 46 |
| 19 | 16 | 164 |
Biochemical activity c-Raf/MEK1/ERK (T202/Y204).
Cellular inhibition assay p-ERK (T202/Y204).
Preliminary results were not encouraging, and weak activity was generally observed for early amides, exemplified by 4-aminobutane-1,2-diol carboxamide (3), which was 103-fold less potent than PD-0325901. The azetidin-3-ol analogue (7) stood out as the most promising starting point for further optimization efforts with a biochemical IC50 of 44 nM and similar potency in the cellular assay. Increasing the ring size of the carboxamide to pyrrolidine or piperidine analogues resulted in a dramatic loss of activity. Oral exposure in the rat was rather poor for (7), the result of low oral availability (19%) and high clearance, and suggested that both absorption and metabolism liabilities need to be addressed going forward (Table 2). Metabolic instability was also reflected in relatively high rat liver microsomal instability observed in vitro. The lipophilic character (cLog P = 4.3, PSA = 52.6 Å2) of 7 suggested further that an improvement in aqueous solubility may also serve to optimize oral exposure.
Table 2. In Vitro ADME and PK Parameters for Lead MEK Inhibitors.
| rat PK parametersa |
|||||||
|---|---|---|---|---|---|---|---|
| compd | MDCKb | RLMc | HLMc | Cl (mL/h/kg) | oral t1/2 (h) | F % | AUC/dosed |
| 7 | 399 | 80.1 | 30.8 | 8324 | 1.5 | 19 | 0.05 |
| 8 | 43 | 16.2 | 755 | 9.1 | 78 | 1.57 | |
| 12 | 207 | 31.5 | 46.3 | 2101 | 5.4 | 101 | 0.85 |
| 13 | 210 | 40.5 | 35.8 | 5958 | 4.9 | 86 | 0.25 |
| 18 | 99 | –4.5 | 2.6 | 800 | 11.4 | 58 | 1.03 |
| 1 | 105 | 22.4 | 13.8 | 3565 | 6.1 | 77 | 0.35 |
Compounds were prepared as amorphous solids and dosed at 5 mg/kg in female CD rats.
Cell permeability Papp nm/s.
% Conversion in the presence of rat and human liver microsomes at 0.5 mg/mL microsomal concentration supplemented with NADPH and at 15 μM substrate concentration at 37 °C for 30 min.
Oral AUC (0–t) normalized to dose (μM h/mg/kg).
The hydroxyl group at the 3-position of the azetidine was important for biochemical potency, and deletion in the case of 6 was poorly tolerated. The cocrystal structure of azetidinol (7) in adenosine 5′-(β,γ-methylenetriphosphonate) (AMP-PCP) bound MEK1 was solved and provided structural insight (Figure 1). The azetidine hydroxyl projects efficiently into the catalytic loop region, forming two hydrogen bonds with residues Asp190 and Asn195 with the diphenylamine core binding in a manner entirely consistent with that of PD-0325901. Azetidin-3-ol interaction with catalytic loop was initially unanticipated and left open the prospect for introduction of additional contacts with bound phosphate and served as the strategic basis for follow up SAR.
On the basis of structural guidance and for pragmatic reasons, our attention was directed primarily to substitution of the azetidine ring 3-position for follow up SAR. We set out to prepare key analogues bearing additional hydrophilic substitutions that could serve to interact with bound ATP phosphate and further enhance aqueous solubility. The introduction of a basic aminomethylene at the 3-position (8) highlighted this effort and led to a significant 6-fold improvement in biochemical activity. Deletion of the basic amine as in the case of 9 resulted in a greater than 10-fold reduction in potency, and nonbasic analogues (10 and 11) failed to improve activity beyond that of 7. The crystal structure of AMP-PCP bound MEK1 with 8 was solved and revealed that the amine does engage the γ-phosphate (Figure 2). The amine appears to induce reorganization of the phosphate chain and brings the groups into close proximity. This shift also allows the alcohol to bridge and engage both Asp190 and the γ-phosphate, resulting in the formation of a more complex network of contacts. Gratifyingly, compound 8 also demonstrated improved oral exposure in the rat relative to 7. Compound 8 was found to have an oral bioavailability of 77% in the rat and a significantly lower clearance, resulting in a dramatic improvement in oral AUC and half-life.
Figure 2.

MEK1:AMP-PCP ternary complex cocrystal structures for 8 (A) and 1 (XL518) (B). Dashed lines indicate key contacts for the carboxamide and aminoethanol fragments.
Compound 8 was examined in a duration of action MDA-MB-231T xenograft PD study that included measurement of BBB penetration and p-ERK inhibition in brain tissue (Table 3). Samples were taken at 2 and 24 h, and at both time points, the concentration of metabolite 4 was measured. At an oral dose of 30 mg/kg, mean plasma and tumor levels of 2.4 and 7.36 μM were observed at 2 h, and some brain tissue exposure was apparent. At 24 h, plasma exposure diminished markedly, and tumor accumulation was observed while brain levels remained relatively unchanged. Minimal 4 was detected in plasma only at 24 h time point and could not be detected in tumor or brain tissue. Overall, the inhibition of p-ERK was modest and apparent in both tumor and brain tissue. While the duration of PD response was encouraging and consistent with sustained tumor exposure, the level of brain tissue activity and an ED50 >30 mg/kg was considered less than ideal for moving forward.
Table 3. PD Activity for Lead MEK Inhibitor (8) and XL518 (1).
| tumorb |
brainb |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| plasmab |
2 h |
24 h |
2 h |
24 h |
|||||||
| compd | dosea | concn 2 h | concn 24 h | concn | p-ERK (%) | concn | p-ERK (%) | concn | p-ERK (%) | concn | p-ERK (%) |
| 8 | 30 | 2.4 | 0.03 | 7.36 | 27 | 26.85 | 44 | 0.73 | 23 | 1.24 | 17 |
| 4c | 0.01 | 0.21 | <LQ | <LQ | <LQ | <LQ | |||||
| 1 | 5 | <LQ | 0.53 | 16 | <LQ | 8 | |||||
| 1 | 10 | 0.1 | 1.99 | 45 | <LQ | 7 | |||||
| 1 | 20 | 0.1 | 3.59 | 75 | <LQ | 2 | |||||
| 1 | 100 | 0.12 | 22.85 | 87 | 0.22 | 0 | |||||
Single oral dosage in MDA-MB-231T tumor bearing mice in mg/kg.
Parent drug and metabolite (4) concentrations given in units of μM. p-ERK (T202/Y204) reported as % inhibition.
Metabolite (4) measured on administration of parent (8).
Going forward, we reasoned that the aminoethanol fragment embedded in 8 should be conserved since it affords optimal contact with ATP γ-phosphate and Asp190. The influence of substitution at the aminomethylene terminus was then explored. A broad set of N-substituted derivatives highlighted by 12–17 were readily synthesized using the same general coupling approach employed for the synthesis of earlier analogues. The required azetidines were prepared straightforwardly from 3-azetidinol (20) (Scheme 2). Mono- and disubstitution of the amine was well tolerated for small groups, and significantly improved cellular activity as well was observed in the case of 12 and 13, although morpholine 17 demonstrated a clear limit to the SAR. Structural studies revealed that nitrogen substitution alters binding by rotation of the amine away from the γ-phosphate to significantly favor interaction with the catalytic loop, presumably driven by steric effects. Rat pharmacokinetic studies revealed relatively poor oral exposure for the series characterized by high clearance and reduced half-life associated in part with metabolic degradation back to parent 8 and correlated well with increased rat microsomal instability in vitro. Methylene substitution was explored by preparation of the individual α-methyl enantiomers (18 and 19). The requisite azetidine precursors were prepared from ethylidine (22) using Sharpless dihydroxylation methodology followed by diol sulfation and regioselective azide ring opening.15 Subsequent azide reduction and protecting group manipulation afforded the desired chiral azetidines (26 and 27). Methylation was well tolerated and the (S)-enantiomer (18) exhibited 3-fold improved potency over the (R)-isomer. Substitution with progressively larger alkyl groups or gem-dialkylation proved unproductive. Compound 18 demonstrated cellular activity and oral exposure analogous to that of the parent methylene 8 and offered no advantage.
Scheme 2. General Synthesis of Azetidine Precursors for Compounds 12–19.

Reagents and conditions: (a) BOC2O, aqueous dioxane, NaHCO3. (b) (COCl)2, DMSO, DCM, −78 °C. (c) RCH2P(Ph)3+Br–, t-BuOK, Et2O, 35 °C. (d) m-CPBA, CHCl3. (e) R1R2NH, THF. (f) TFA. (g) AD-mix-β, MeSO2NH2, aqueous t-BuOH. (h) (i) HCl, MeOH; (ii) CbzCl, aqueous dioxane, NaHCO3. (i) SOCl2, pyr., DCM, 0 °C. (j) RuCl3, NaIO4, aqueous ACN. (k) NaN3, DMF, 60 °C. (l) PPh3, aqueous THF, 70 °C. (m) BOC2O, THF. (n) Pd/C (10%), H2, MeOH, 1 atm.
The strategy for final optimization recognized the opportunity to combine the tolerance for N-alkylation and α-methylene substitution. Ultimately, the ideal balance of optimal cellular potency and oral exposure was achieved by linking the methylene and terminal nitrogen through an alicyclic ring. A piperidine ring was found to be optimal and on the laboratory scale was isolated in chiral form by resolution of 28, prepared according to the published method of Peter Beak (Scheme 3).16 The piperidin-2-yl (S)-isomer (1) (XL518, GDC- 0973) was found to be over 10-fold more potent than the (R)-isomer and exhibits subnanomolar biochemical and cellular activity. The cocrystal structure of XL518 in AMP-PCP bound MEK1 was solved and revealed the formation of a highly complex network of interactions involving the embedded aminoethanol terminus with both the catalytic loop Asp190 and the γ-phosphate (Figure 2). Introduction of the cyclic constraint repositions the amine closer to the catalytic loop and allows for engagement of both γ-phosphate and Asp190. Acceptable oral exposure in the rat was observed with half-life and clearance values intermediate that of the N-substituted derivative (13) and parent (8), which translated into potent PD activity as well (Table 3). In a dose ranging duration of action PD study, limited brain exposure was observed at the 100 mg/kg dose, and no metabolite (4) could be detected. An ED90 of 39.7 mg/kg for p-ERK inhibition in tumor tissue at 24 h was measured and provided support for an improved safety profile of 1 in terms of minimizing MAPK pathway inhibition in brain tissue within an effective therapeutic dose range. In an MDA-MB-231T efficacy study, XL518 demonstrated tumor growth inhibition values of 60 and 93% at 1 and 3 mg/kg, respectively, and statistically significant tumor regression was observed at higher doses.17 Overall, predicted ED50 and ED90 values based on this study are 0.6 and approximately 3 mg/kg/day, respectively, in the latter case corresponding to peak circulating plasma levels in the range of 130 nM. Intermediate serum protein binding in the range of 94–96% across species as well as a disproportionate accumulation of XL518 into the tumor tissue in xenograft studies affords a disconnect between effective drug concentrations under serum-free conditions in vitro with those in vivo. XL518 demonstrated no mutagenicity in Ames testing and was well tolerated in subchronic oral dosing studies in rats with minimal effects on plasma clinical chemistry and hematology parameters.
Scheme 3. Resolution of Piperdines for the Synthesis of XL518.

Reagents and conditions: (a) (i) (R)-(−)-α-Methoxy-α-trifluoromethylphenylacetyl chloride, DMAP, DCM, 0 °C; (ii) Silica gel chromatography. (b) Aqueous NaOH, MeOH. (c) Pd/C (10%), H2, MeOH, 1 atm.
In summary, a series of highly potent and selective MEK inhibitors based on the diphenylamine core have been identified. This novel series makes use of a metabolically stable azetidinyl carboxamide linkage that exploits unique and efficient contacts with bound ATP γ-phosphate and Asp190 in the catalytic loop region. Efforts to optimize the potency and pharmacokinetic profile of this series culminated in the discovery of the XL518 (GDC-0973). XL518 demonstrates excellent pharmacokinetics in preclinical species and minimal BBB penetration by parent or active metabolite (4) that translates into sustained tumor tissue selective inhibition of the ERK/MAPK pathway in PD studies. Potent in vivo efficacy in tumor xenograft models using a qd dosing regimen compares favorably with PD-0325901. Early preclinical safety assessments indicate that XL518 is well tolerated when dosed subchronically to achieve efficacious circulating concentrations and is currently in early clinical development as both a single agent and in combination with the class I PI3K inhibitor GDC-0941.
Acknowledgments
We thank John Woolfrey, Lester Bornheim, and Jae Lee for project team support, Paul Foster for structural and graphics support, Siamak Dailami for analytical and instrumentation support, and Tom Von Geldern and Stuart McCombie for helpful discussions.
Supporting Information Available
Experimental procedures for the synthesis and characterization of 1 and 6–19, MEK1 cocrystal structure determinations, kinase selectivity data for XL518, and details for key in vitro and in vivo studies. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Luo X.; Ruhland M. K.; Pazolli E.; Lind A. C.; Stewart S. A Osteopontin Stimulates Preneoplastic Cellular Proliferation Through Activation of the MAPK Pathway. Mol. Cancer Res. 2011, 981018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand M.; Meter T. E.; Fillmore H. L. Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1) expression and invasion in glioma cell lines via the MAPK pathway. J. Neuro-Oncol. 2011, 1043679–687. [DOI] [PubMed] [Google Scholar]
- Romon R.; Adriaenssens E.; Lagadec C.; Germain E.; Hondermarck H.; Le Bourhis X. Nerve growth factor promotes breast cancer angiogenesis by activating multiple pathways. Mol. Cancer 2010, 9, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloster M. M.; Naderi E. H.; Carlsen H.; Blomhoff H. K.; Naderi S. Hyperactivation of NF-κ B via the MEK signaling is indispensable for the inhibitory effect of cAMP on DNA damage-induced cell death. Mol. Cancer 2011, 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Medarde A.; Santos E. Ras in cancer and developmental diseases. Genes Cancer 2011, 23344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matallanas D.; Birtwistle M.; Romano D.; Zebisch A.; Rauch J.; von Kriegsheim A.; Kolch W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 23232–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin A. G.; Bleam M. R.; Groy A.; Moss K. G.; Minthorn E. A.; Kulkarni S. G.; Rominger C. M.; Erskine S.; Fisher K. E.; Yang J.; Zappacosta F.; Annan R.; Sutton D.; Laquerre S. G. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 2011, 175989–1000. [DOI] [PubMed] [Google Scholar]
- Bennouna J.; Lang I.; Valladares-Ayerbes M.; Boer K.; Adenis A.; Escudero P.; Kim T.-Y.; Pover G. M.; Morris C. D.; Douillard J.-Y. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest. New Drugs 2011, 2951021–1028. [DOI] [PubMed] [Google Scholar]
- Barrett S. D.; Bridges A. J.; Dudley D. T.; Saltiel A. R.; Fergus J. H.; Flamme C. M.; Delaney A. M.; Kaufman M.; LePage S.; Leopold W. R.; Przybranowski S. A.; Sebolt-Leopold J.; Van Becelaere K.; Doherty A. M.; Kennedy R. M.; Marston D.; Howard W. A. Jr.; Smith Y.; Warmus J. S. Tecle Haile The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD-0325901. Bioorg. Med. Chem. Lett. 2008, 18246501–6504. [DOI] [PubMed] [Google Scholar]
- Mirzoeva O. K.; Das D; Heiser L. M. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009, 692565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich K. P.; Merchant M.; Orr C.; Chan J.; Den Otter D.; Berry L.; Kasman I.; Koeppen H.; Rice K.; Yang N. Y.; Engst S.; Johnston S.; Friedman L. S.; Belvin M. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012, 721210–219. [DOI] [PubMed] [Google Scholar]
- Wabnitz P. A.; Mitchell D.; Wabnitz D. A. M. In Vitro and In Vivo Metabolism of the Anti-Cancer Agent CI-1040, a MEK Inhibitor, in Rat, Monkey, and Human. Pharm. Res. 2004, 2191670–1679. [DOI] [PubMed] [Google Scholar]
- Haura E. B.; Ricart A. D.; Larson T. G.; Stella P. J.; Bazhenova L.; Miller V. A.; Cohen R. B.; Eisenberg P. D.; Selaru P.; Wilner K. D.; Gadgeel S. M. A Phase II Study of PD-0325901, an Oral MEK Inhibitor, in Previously Treated Patients with Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2010, 1682450–2457. [DOI] [PubMed] [Google Scholar]
- Ohren J. F.; Chen H.; Pavlovsky A.; Whitehead C.; Zhang E.; Kuffa P.; Yan C.; McConnel P.; Spressard C.; Banotai C.; Mueller W. T.; Delaney A.; Omer C.; Sebolt-Leopold J.; Dudley D.; Leung I.; Flamme C.; Warmus J. S.; Kaufman M.; Barrett S.; Tecle H.; Hasemann C. Nat. Struct. Mol. Biol. 2004, 11, 1192. [DOI] [PubMed] [Google Scholar]
- Sharpless K. B.; Amberg W.; Bennani Y. L.; Crispino G. A.; Hartung J.; Jeong K.-S.; Kwong H.-L.; Morikawa K.; Wang Z.-M.; Xu D.; Zhang X.-L. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768. [Google Scholar]
- Beak P.; Lee W. K. α-Lithioamine synthetic equivalents: syntheses of diastereoisomers from the Boc-piperidines. J. Org. Chem. 1990, 5592578–80. [Google Scholar]
- Johnston S.XL518, a potent selective orally bioavailable MEK1 inhibitor, downregulates the RAS/Raf/MEK/ERK pathway in vivo, resulting in tumor growth inhibition and regression in pre-clinical models. AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. Oct. 22–26, 2007, San Francisco, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.