

Abstract

MurD and MurE ligases, consecutive enzymes participating in the intracellular steps of bacterial peptidoglycan biosynthesis, are important targets for antibacterial drug discovery. We have designed, synthesized, and evaluated the first d-glutamic acid-containing dual inhibitor of MurD and MurE ligases from Escherichia coli and Staphylococcus aureus (IC50 values between 6.4 and 180 μM) possessing antibacterial activity against Gram-positive S. aureus and its methicillin-resistant strain (MRSA) with minimal inhibitory concentration (MIC) values of 8 μg/mL. The inhibitor was also found to be noncytotoxic for human HepG2 cells at concentrations below 200 μM.
Keywords: antibacterial agent, inhibitor, Mur ligase, peptidoglycan, 2-thioxothiazolidin-4-one
The increasing emergence of bacterial strains resistant to currently available antibiotics has created unmet medical needs in antibacterial therapy. Development of novel effective antibacterial drugs directed toward previously unexploited targets is therefore essential to combat bacterial drug resistance in the future.1 Peptidoglycan, an essential cell wall polymer unique to prokaryotic cells, is thus an important target for antibacterial drug discovery.2 The biosynthesis of peptidoglycan is a multistep process comprising intracellular assembly of the uridine 5′-diphosphate (UDP)-MurNAc-pentapeptide (Park's nucleotide), which is subsequently translocated through the cytoplasmic membrane and incorporated into the growing peptidoglycan layer.3 While the majority of currently approved antibiotics that target peptidoglycan biosynthesis act on the extracellular transpeptidation steps,4 there has been increased interest in exploiting the enzymes involved in the early intracellular steps of peptidoglycan precursor biosynthesis.5 This is in particular the case for ATP-dependent Mur ligases [UDP-N-acetylmuramate:l-Ala ligase (MurC) to UDP-N-acetylmuramoyl-l-Ala-γ-d-Glu-meso-diaminopimelate(or l-Lys):d-Ala-d-Ala ligase (MurF)], which catalyze a series of reactions leading to Park's nucleotide by sequentially adding l-Ala (MurC), d-Glu [UDP-N-acetylmuramoyl-l-Ala:d-Glu ligase (MurD)], l-Lys or meso-diaminopimelic acid [UDP-N-acetylmuramoyl-l-Ala-d-Glu:meso-diaminopimelate(or l-Lys) ligase (MurE)], and d-Ala-d-Ala dipeptide (MurF) to the starting MurC substrate UDP-MurNAc.3
Mur ligases catalyze the formation of a peptide or amide bond between the UDP-substrate and the condensing amino acid. They operate by similar chemical mechanisms6,7 and, as shown for MurC and MurF, by an ordered kinetic mechanism.8,9 The enzymatic reaction is initiated by the binding of ATP to the free enzyme, followed by binding of the corresponding UDP substrate. The terminal carboxyl group of the UDP substrate is then activated by ATP-promoted phosphorylation, resulting in the formation of an acylphosphate intermediate that is attacked by the amino group of the incoming amino acid residue or dipeptide after its binding to the enzyme. The resulting tetrahedral high-energy intermediate collapses with elimination of inorganic phosphate and concomitant formation of a peptide or amide bond.6−9 The crystal structures of Mur ligases from different bacterial strains show the similar three-domain topology, with the N-terminal and central domains binding UDP precursor and ATP, respectively, while the C-terminal domain binds the condensing amino acid or dipeptide residue. While the topologies of the central and C-terminal domains are similar among the Mur ligases, those of the N-terminal domains show differences, with MurC and MurD more closely related to each other than to MurE and MurF. These differences are related to the lengths of the UDP precursor substrates.10
Recently, we have designed a series of thiazolidin-4-one-based inhibitors of Mur ligases that act either as multitarget11,12 or MurD-selective inhibitors.13−16 Multitarget inhibitors of bacterial enzymes constitute a promising strategy to combat bacterial resistance because target-mediated resistance to such compounds is less likely to evolve, since mutations conferring resistance would have to occur in at least two different target genes during a single generation.17 Although sequence alignment of Mur ligase orthologues and paralogues revealed relatively low overall homology, there is quite high homology of residues present especially in the ATP-binding site.18−21 The common catalytic mechanism and the conserved active sites make Mur ligases attractive targets for the design of multitarget inhibitors.11,12,22,23 Because MurD and MurE ligases are consecutive enzymes in the biosynthesis of Park's nucleotide (Figure S1 in the Supporting Information), it is reasonable to assume that it would be possible to inhibit both enzymes with a single molecule that would act as MurD product and MurE substrate analogue. Moreover, superposition of MurD and MurE crystal structures revealed that residues important for binding of d-Glu in the MurD enzyme have their corresponding counterparts in the MurE enzyme, which gives further support to our hypothesis.22 Indeed, glutamic acid-based MurD inhibitors, designed as transition-state analogues, were reported to inhibit Staphylococcus aureus MurE to some extent.24,25 Similarly, compound I (Figure 1) and its analogous Escherichia coli MurD inhibitors showed weak affinity for S. aureus MurE with residual activities (RA) between 35 and 76% at 500 μM compound (Table S1 in the Supporting Information). Although reported MurE inhibitions are in general moderate, these observations provide the rationale for evaluating MurD inhibitors for MurE inhibition.
Figure 1.
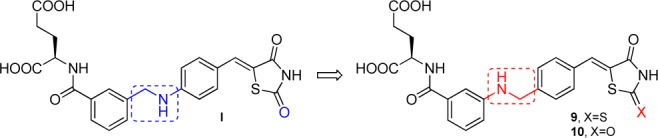
Structural modification of E. coli MurD inhibitor I.
Analysis of the binding mode of previously reported thiazolidine-2,4-dione-based inhibitor I (Figure 1) in the E. coli MurD active site revealed that the d-Glu moiety and the thiazolidine-2,4-dione ring form the majority of hydrogen bonds with E. coli MurD active site residues, while the linker between them is involved mainly in hydrophobic interactions and hydrogen bonds through water molecules.14,15 Compound I inhibited E. coli MurD ligase with an IC50 of 35 μM14 and also showed weak inhibition of S. aureus MurE ligase (RA = 64% at 500 μM, Table 1). Using structure-based design, through several structure optimization cycles, we improved E. coli MurD ligase inhibitory potency of I down to an IC50 value of 3 μM.15 Here, we report further structural modifications of the E. coli MurD inhibitor I by variation of the linker connecting the two phenyl rings and replacement of the thiazolidine-2,4-dione ring by the rhodanine moiety (Figure 1), while leaving other parts of the molecule unchanged, given their important interactions with E. coli MurD active site residues. The synthesis of target compounds 9 and 10 is outlined in Scheme 1 and described in detail in the Supporting Information.
Table 1. Inhibitory Activity of Compounds 9 and 10 against MurD and MurE Ligases and Their Antibacterial Activity.
| IC50 (μM) |
MIC (μg/mL) |
|||||
|---|---|---|---|---|---|---|
| enzyme | I | 9 | 10 | bacterial strain | 9 | 10 |
| E. coli MurD ligase | 35 | 8.2 ± 0.6 | 36 ± 5 | S. aureus ATCC 29213 | 8 | >128 |
| S. aureus MurD ligase | ND | 6.4 ± 1.0 | 100 ± 5 | MRSA ATCC 43300 | 8 | >128 |
| E. coli MurE ligase | 64%a | 180 ± 60 | ∼200b | E. faecalis ATCC 29212 | 64 | >128 |
| S. aureus MurE ligase | ND | 17 ± 1.5 | >200c | E. coli ATCC 25922 | >128 | >128 |
| P. aeruginosa ATCC 27853 | >128 | >128 | ||||
Residual activity (RA) at 500 μM compound.
Residual activity at 200 μM compound: 51 ± 2%.
No inhibitory activity at 200 μM compound; ND, not determined.
Scheme 1. Synthesis of Target Compounds 9 and 10.

Reagents and conditions: (a) Ethylene glycol, p-toluenesulfonic acid, benzene, reflux, 24 h. (b) 3-Aminobenzoic acid, NaCNBH3, MeOH, rt, 16 h. (c) Pyridinium tosylate, THF/H2O, rt, 18 h. (d) Compound 5: rhodanine, piperidine, AcOH, EtOH, reflux, 48 h; compound 6: thiazolidine-2,4-dione, piperidine, AcOH, EtOH, microwave, 140 °C, 30 min. (e) H-d-Glu(OMe)-OMe·HCl, O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU), Et3N, CH2Cl2, rt, 5 h. (f) Compound 9: 2 M LiOH, MeOH/H2O, rt, 16 h; compound 10: 1 M NaOH, MeOH/H2O, rt, 3 h.
Considering a possibility of dual MurD and MurE inhibition, the target compounds 9 and 10 were tested against MurD and MurE ligases from E. coli and S. aureus using the radioactivity assays (Table 1). The compounds were tested in the presence of detergent Tween-20 to avoid nonspecific inhibition due to aggregate formation.26 Compound 9 inhibited MurD ligases from E. coli and S. aureus with IC50 values of 8.2 and 6.4 μM, respectively, and additionally showed inhibitory activity against MurE ligases from E. coli and S. aureus with IC50 values of 180 and 17 μM, respectively, thus acting as a dual inhibitor of the intracellular steps of peptidoglycan biosynthesis. Dual inhibition of MurD and MurE from S. aureus was well balanced, which was not the case for MurD and MurE from E. coli. Thiazolidine-2,4-dione 10 was found to be weaker inhibitor of all four Mur ligases than its rhodanine counterpart 9, which has already been observed previously in the case of E. coli MurD for similar compounds.13−16 Compound 10 inhibited MurD ligases from E. coli and S. aureus with IC50 values of 36 and 100 μM, respectively, while it was found to be inactive against S. aureus MurE and showed comparable E. coli MurE inhibition as 9.
Following the promising inhibition of MurD and MurE ligases from the two bacterial strains, inhibitors 9 and 10 were also tested for their antibacterial activity against three Gram-positive [S. aureus ATCC 29213, methicillin-resistant S. aureus (MRSA) ATCC 43300, and Enterococcus faecalis ATCC 29212] and two Gram-negative (E. coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853) bacteria (Table 1). Although compound 9 was found to be inactive against both Gram-negative bacteria, presumably because of its inability to cross the outer membrane, it inhibited the growth of E. faecalis with MIC of 64 μg/mL and showed more potent activity against S. aureus and MRSA with MIC values of 8 μg/mL. Compound 10, a weaker inhibitor of Mur ligases than 9, was devoid of antibacterial activity. Compound 9 represents, to the best of our knowledge, the first d-Glu-based inhibitor of Mur ligases possessing antibacterial activity; however, its mode of action still remains to be determined. To evaluate the preliminary safety profile of compound 9, it was tested for its cytotoxicity on human HepG2 cells and found to be noncytotoxic at concentrations at least up to 200 μM (Figure S2 in the Supporting Information).
The binding mode of inhibitor 9 in the E. coli MurD active site was determined by solving the crystal structure of E. coli MurD–9 complex, the structure of E. coli apo-MurD being employed as a search model in a molecular replacement experiment (Figures 2 and S3 in the Supporting Information). In the crystal structure of E. coli MurD–9 complex, solved to a resolution of 1.5 Å, the inhibitor is seen to occupy the binding site of MurD product UDP-MurNAc-l-Ala-d-Glu (UMAG). The d-Glu moiety of the inhibitor is positioned at the same site as that previously observed for the d-Glu residue of UMAG6 and other MurD inhibitors containing the d-Glu moiety,14,15,27,28 while the rhodanine moiety occupies the uracil-binding pocket, where it forms a hydrogen bond with Oγ of Thr36. However, there are important differences in the position of α-carboxylate group of d-Glu moiety (Figures 2 and S4 in the Supporting Information). The α-carboxylate group of the d-Glu moiety of UMAG and of the previously reported thiazolidin-4-one inhibitor I forms a charge-based interaction with Nξ of Lys348 and is additionally hydrogen-bonded through a conserved water molecule to Oγ of Thr321 and the carboxyl group of Asp182. In the case of E. coli MurD–9 complex, the α-carboxylate group of d-Glu is rotated by approximately 75°, which results in a different hydrogen-bonding network. Because of a different overall conformation, the α-carboxylate group loses direct contact with the Nξ of Lys348 and forms hydrogen bonds with three water molecules (W19, W92, and W102), which form a hydrogen bond network with residues Lys115, Glu157, Asp182, Thr321, and Lys348 (for more details, see Figure S3 and the text in the Supporting Information). The described linker modification of I to give 9 results in the different conformation of the middle part of the inhibitors (Figure S4 in the Supporting Information). The benzylidene moiety of 9 is held in place by the hydrophobic interaction of the phenyl ring with Gly73, while the phenyl ring of the phenylamino moiety contributes to recognition of the inhibitor within the active site, with hydrophobic interactions to Leu416 and possible π–π interactions to Phe161. In addition to these hydrophobic interactions, the most crucial interactions between the inhibitor 9 and E. coli MurD active site residues are hydrogen bonds formed between the carboxylates of d-Glu moiety and Thr321, Lys348, Ser415, and Phe422, since they contribute to the recognition of the d-Glu moiety in the d-Glu-binding site, as well as hydrogen bond between rhodanine NH group and Thr36. On the other hand, it is hard to estimate the importance of the water-mediated interactions, since it was shown by high-resolution NMR and molecular dynamics that the bound ligands exhibit conformational flexibility in the E. coli MurD active site.29 Therefore, water-mediated interactions in the solution are very different from those observed in the crystal structure.
Figure 2.

X-ray binding mode of inhibitor 9 (in magenta) in the active site of E. coli MurD ligase (PDB code: 2Y1O). Hydrogen bonds between 9 and the E. coli MurD active site residues (in yellow) and crystal water molecules (in red) are presented as dashed green lines. Comparison of the binding modes of inhibitors I and 9 in the E. coli MurD active site is shown in Figure S4 in the Supporting Information.
In summary, structural modification of our recently disclosed thiazolidin-4-one-based MurD inhibitors led to the discovery of compound 9 that inhibited E. coli and S. aureus MurD ligase with IC50 values of 8.2 and 6.4 μM, respectively. It also inhibited E. coli and S. aureus MurE ligases with respective IC50 values of 180 and 17 μM. Compound 9 is, to the best of our knowledge, the first d-Glu-based dual inhibitor of Mur ligases that possesses antibacterial activity against Gram-positive S. aureus and MRSA with MIC values of 8 μg/mL; however, its mode of action still remains to be determined. Moreover, compound 9 was found to be noncytotoxic on human HepG2 cells at concentrations at least up to 200 μM. These results make compound 9 a promising inhibitor of two consecutive steps of peptidoglycan biosynthesis and highlight it as an interesting candidate for further optimization toward compounds with improved antibacterial activity.
Acknowledgments
We thank Professor Roger Pain for critical reading of the manuscript.
Glossary
Abbreviations
- UDP
uridine 5′-diphosphate
- UMA
UDP-N-acetylmuramoyl-l-Ala
- UMAG
UDP-N-acetylmuramoyl-l-Ala-d-Glu
- MurC
UDP-N-acetylmuramate:l-Ala ligase
- MurD
UDP-N-acetylmuramoyl-l-Ala:d-Glu ligase
- MurE
UDP-N-acetylmuramoyl-l-Ala-d-Glu:meso-diaminopimelate(or l-Lys) ligase
- MurF
UDP-N-acetylmuramoyl-l-Ala-γ-d-Glu-meso-diaminopimelate(or l-Lys):d-Ala-d-Ala ligase
Supporting Information Available
Experimental procedures and characterization of intermediate and target compounds, description of crystallization, preparation of the inhibitor complex, data collection, crystal structure solution, and model refinement, description of enzyme assays, and determination of antibacterial activity and cytotoxicity assay. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The PDB code for the E. coli MurD–9 complex is 2Y1O.
This work was supported by the EU FP6 Integrated Project EUR-INTAFAR (Project No. LSHM-CT-2004-512138), by the Slovenian Research Agency (Grant No. P1-0208), and by the World Federation of Scientists.
The authors declare no competing financial interest.
Supplementary Material
References
- Silver L. L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W.; Blanot D.; de Pedro M. A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [DOI] [PubMed] [Google Scholar]
- Barreteau H.; Kovač A.; Boniface A.; Sova M.; Gobec S.; Blanot D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 168–207. [DOI] [PubMed] [Google Scholar]
- Green D. W. The bacterial cell wall as a source of antibacterial targets. Expert Opin. Ther. Targets 2002, 6, 1–19. [DOI] [PubMed] [Google Scholar]
- El Zoeiby A.; Sanschagrin F.; Levesque R. C. Structure and function of the Mur enzymes: Development of novel inhibitors. Mol. Microbiol. 2003, 47, 1–12. [DOI] [PubMed] [Google Scholar]
- Bertrand J. A.; Auger G.; Martin L.; Fanchon E.; Blanot D.; Le Beller D.; van Heijenoort J.; Dideberg O. Determination of the MurD mechanism through crystallographic analysis of enzyme complexes. J. Mol. Biol. 1999, 289, 579–590. [DOI] [PubMed] [Google Scholar]
- Bouhss A.; Dementin S.; van Heijenoort J.; Parquet C.; Blanot D. MurC and MurD synthetases of peptidoglycan biosynthesis: borohydride trapping of acyl-phosphate intermediates. Methods Enzymol. 2002, 354, 189–196. [DOI] [PubMed] [Google Scholar]
- Emanuele J. J. Jr.; Jin H.; Yanchunas J. Jr.; Villafranca J. J. Evaluation of the kinetic mechanism of Escherichia coli uridine diphosphate-N-acetylmuramate:L-alanine ligase. Biochemistry 1997, 36, 7264–7271. [DOI] [PubMed] [Google Scholar]
- Anderson M. S.; Eveland S. S.; Onishi H. R.; Pompliano D. L. Kinetic mechanism of the Escherichia coli UDPMurNAc-tripeptide D-alanyl-D-alanine-adding enzyme: Use of a glutathione S-transferase fusion. Biochemistry 1996, 35, 16264–16269. [DOI] [PubMed] [Google Scholar]
- Smith C. A. Structure, function and dynamics in the mur family of bacterial cell wall ligases. J. Mol. Biol. 2006, 362, 640–655. [DOI] [PubMed] [Google Scholar]
- Tomašić T.; Zidar N.; Kovač A.; Turk S.; Simčič M.; Blanot D.; Müller-Premru M.; Filipič M.; Grdadolnik S. G.; Zega A.; Anderluh M.; Gobec S.; Kikelj D.; Peterlin Mašič L. 5-Benzylidenethiazolidin-4-ones as multitarget inhibitors of bacterial Mur ligases. ChemMedChem 2010, 5, 286–295. [DOI] [PubMed] [Google Scholar]
- Tomašić T.; Kovač A.; Klebe G.; Blanot D.; Gobec S.; Kikelj D.; Peterlin Mašič L. Virtual screening for potential inhibitors of bacterial MurC and MurD ligases. J. Mol. Model. 2012, 18, 1063–1072. [DOI] [PubMed] [Google Scholar]
- Tomašić T.; Zidar N.; Rupnik V.; Kovač A.; Blanot D.; Gobec S.; Kikelj D.; Peterlin Mašič L. Synthesis and biological evaluation of new glutamic acid-based inhibitors of MurD ligase. Bioorg. Med. Chem. Lett. 2009, 19, 153–157. [DOI] [PubMed] [Google Scholar]
- Zidar N.; Tomašić T.; Šink R.; Rupnik V.; Kovač A.; Turk S.; Patin D.; Blanot D.; Martel C. C.; Dessen A.; Müller-Premru M.; Zega A.; Gobec S.; Peterlin Mašič L.; Kikelj D. Discovery of novel 5-benzylidenerhodanine and 5-benzylidenethiazolidine-2,4-dione inhibitors of MurD ligase. J. Med. Chem. 2010, 53, 6584–6594. [DOI] [PubMed] [Google Scholar]
- Tomašić T.; Zidar N.; Šink R.; Kovač A.; Blanot D.; Contreras-Martel C.; Dessen A.; Müller-Premru M.; Zega A.; Gobec S.; Kikelj D.; Peterlin Mašič L. Structure-based design of a new series of D-glutamic acid based inhibitors of bacterial UDP-N-acetylmuramoyl-L-alanine:D-glutamate ligase (MurD). J. Med. Chem. 2011, 54, 4600–4610. [DOI] [PubMed] [Google Scholar]
- Tomašić T.; Kovač A.; Simčič M.; Blanot D.; Grdadolnik S. G.; Gobec S.; Kikelj D.; Peterlin Mašič L. Novel 2-thioxothiazolidin-4-one inhibitors of bacterial MurD ligase targeting D-Glu- and diphosphate-binding sites. Eur. J. Med. Chem. 2011, 46, 3964–3975. [DOI] [PubMed] [Google Scholar]
- Wong K. K.; Kuo D. W.; Chabin R. M.; Fournier C.; Gegnas L. D.; Waddell S. T.; Marsilio F.; Leiting B.; Pompliano D. L. Engineering a cell-free murein biosynthetic pathway: Combinatorial enzymology in drug discovery. J. Am. Chem. Soc. 1998, 120, 13527–13528. [Google Scholar]
- Ikeda M.; Wachi M.; Jung H. K.; Ishino F.; Matsuhashi M. Homology among MurC, MurD, MurE and MurF proteins in Escherichia coli and that between Escherichia coli MurG and a possible MurG protein in Bacillus subtilis. J. Gen. Appl. Microbiol. 1990, 36, 179–187. [Google Scholar]
- Bouhss A.; MenginLecreulx D.; Blanot D.; van Heijenoort J.; Parquet C. Invariant amino acids in the mur peptide synthetases of bacterial peptidoglycan synthesis and their modification by site-directed mutagenesis in the UDP-MurNAc:L-alanine ligase from Escherichia coli. Biochemistry 1997, 36, 11556–11563. [DOI] [PubMed] [Google Scholar]
- Eveland S. S.; Pompliano D. L.; Anderson M. S. Conditionally lethal Escherichia coli murein mutants contain point defects that map to regions conserved among murein and folyl poly-gamma-glutamate ligases: Identification of a ligase superfamily. Biochemistry 1997, 36, 6223–6229. [DOI] [PubMed] [Google Scholar]
- Bouhss A.; Dementin S.; Parquet C.; Mengin-Lecreulx D.; Bertrand J. A.; Le Beller D.; Dideberg O.; van Heijenoort J.; Blanot D. Role of the ortholog and paralog amino acid invariants in the active site of the UDP-MurNAc-L-alanine:D-glutamate ligase (MurD). Biochemistry 1999, 38, 12240–12247. [DOI] [PubMed] [Google Scholar]
- Perdih A.; Kovač A.; Wolber G.; Blanot D.; Gobec S.; Šolmajer T. Discovery of novel benzene 1,3-dicarboxylic acid inhibitors of bacterial MurD and MurE ligases by structure-based virtual screening approach. Bioorg. Med. Chem. Lett. 2009, 19, 2668–2673. [DOI] [PubMed] [Google Scholar]
- Sova M.; Kovač A.; Turk S.; Hrast M.; Blanot D.; Gobec S. Phosphorylated hydroxyethylamines as novel inhibitors of the bacterial cell wall biosynthesis enzymes MurC to MurF. Bioorg. Chem. 2009, 37, 217–222. [DOI] [PubMed] [Google Scholar]
- Humljan J.; Kotnik M.; Boniface A.; Šolmajer T.; Urleb U.; Blanot D.; Gobec S. A new approach towards peptidosulfonamides: Synthesis of potential inhibitors of bacterial peptidoglycan biosynthesis enzymes MurD and MurE. Tetrahedron 2006, 62, 10980–10988. [Google Scholar]
- Štrancar K.; Boniface A.; Blanot D.; Gobec S. Phosphinate inhibitors of UDP-N-acetylmuramoyl-L-alanyl-D-glutamate:L-lysine ligase (MurE). Arch. Pharm. (Weinheim) 2007, 340, 127–134. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Helfand B. T.; Feng B.; Shoichet B. K. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003, 46, 4265–4272. [DOI] [PubMed] [Google Scholar]
- Kotnik M.; Humljan J.; Contreras-Martel C.; Oblak M.; Kristan K.; Hervé M.; Blanot D.; Urleb U.; Gobec S.; Dessen A.; Šolmajer T. Structural and functional characterization of enantiomeric glutamic acid derivatives as potential transition state analogue inhibitors of MurD ligase. J. Mol. Biol. 2007, 370, 107–115. [DOI] [PubMed] [Google Scholar]
- Humljan J.; Kotnik M.; Contreras-Martel C.; Blanot D.; Urleb U.; Dessen A.; Šolmajer T.; Gobec S. Novel naphthalene-N-sulfonyl-D-glutamic acid derivatives as inhibitors of MurD, a key peptidoglycan biosynthesis enzyme. J. Med. Chem. 2008, 51, 7486–7494. [DOI] [PubMed] [Google Scholar]
- Simčič M.; Hodošček M.; Humljan J.; Kristan K.; Urleb U.; Kocjan D.; Golič Grdadolnik S. NMR and molecular dynamics study of the binding mode of naphthalene-N-sulfonyl-D-glutamic acid derivatives: novel MurD ligase inhibitors. J. Med. Chem. 2009, 52, 2899–2908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.