

Abstract

β-Secretase inhibitors are potentially disease-modifying treatments for Alzheimer's disease. Previous efforts in our laboratory have resulted in hydroxyethylamine-derived inhibitors such as 1 with low nanomolar potency against β-site amyloid precursor protein cleaving enzyme (BACE). When dosed intravenously, compound 1 was also shown to significantly reduce Aβ40 levels in plasma, brain, and cerebral spinal fluid. Herein, we report further optimizations that led to the discovery of inhibitor 16 as a novel, potent, and orally efficacious BACE inhibitor.
Keywords: β-site amyloid precursor protein cleaving enzyme (BACE), Alzheimer's disease (AD), hydroxyethylamine (HEA) isostere
The need for a disease-modifying treatment for Alzheimer's disease (AD) continues to grow. In the United States alone, the number of cases exceeds 5 million, and AD is the seventh leading cause of death.1 The disease is characterized by the progressive loss of cognitive ability, dementia, and eventually death. Extracelullar amyloid plaques, largely comprised of Aβ40–42 peptides, and intracellular neurofibrillary tangles are the defining pathological characteristics of AD. Aβ40–42 is a product of the cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase. The cloning of the β-site amyloid precursor protein cleaving enzyme (BACE) enzyme and its critical role in the amyloid cascade2 have led to the hypothesis that BACE inhibitors are potential disease-modifying treatments for AD.3−3c During the past decade, numerous pharmaceutical companies have committed considerable resources to develop BACE inhibitors.4,4b In this letter, we will describe our effort toward the design and synthesis of a novel, potent, and orally efficacious BACE inhibitor.
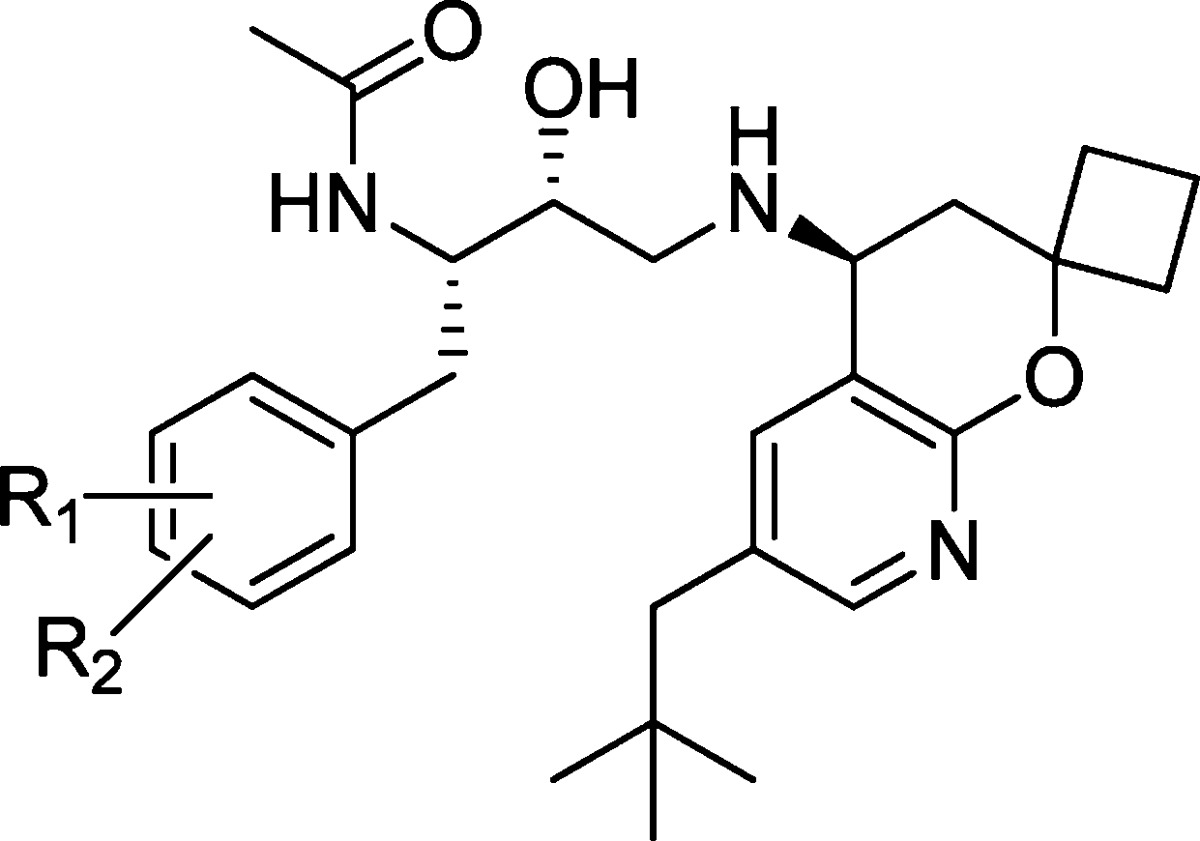
Previous work in our laboratories revealed hydroxyethylamine (HEA)-derived compound 1 (Figure 1) to be a potent BACE inhibitor.5 On the basis of X-ray analysis, the HEA transition state isostere binds to the two catalytic aspartyl residues in the expected fashion. The N-cyclopentyl-3-(2-pyridyl) pyridone amide moiety of 1 provides interactions with the S2 and S3 binding pockets of BACE, and the 6-ethyl 2,2-spirocyclobutylchromane engages in hydrophobic interactions with the S2′ and S1′ binding pockets. Compound 1 exhibited potent cellular activity (8.8 nM); however, it also displayed poor metabolic stability (mouse liver microsomes = 568 μL/min/mg), and its measured in vivo clearance was greater than liver blood flow (mouse iv, 2 mg/kg, 5.6 L/h/kg). Despite these liabilities, when dosed intravenously in Tg2576 transgenic mice6 (30 mg/kg, 4 h time point), compound 1 significantly reduced Aβ40 levels by 91, 58, and 57% in plasma, brain, and cerebral spinal fluid (CSF), respectively.
Figure 1.

Truncating the amide of 1 and optimizing P2′ led to BACE inhibitor 4 (S1–S2′ indicate BACE binding domains).
Although compound 1 displays impressive enzymatic inhibition, it is not an efficient inhibitor of BACE (BE = 0.24)7 and has a relatively high molecular weight of 647 amu. It also has a large total polar surface area (PSA), high clog P, and the number of H-bond donors and acceptors are not ideal for a target located in the CNS.8 To optimize the properties of 1, guided by the well-established binding models, we reasoned that the HEA core had to be kept intact to properly engage the catalytic aspartic residues of BACE. We decided to target the pyridone and chromane moieties of 1 as the most likely regions to provide meaningful changes to the physiochemical properties of 1, while maintaining high cellular potency against BACE. A major advancement in our early structure–activity relationship (SAR) was the identification of the highly truncated acetamide analogue 2.9 Despite the removal of P2 and P3 interactions with the enzyme, acetamide 2 retained modest activity but with higher binding efficiency (BE = 0.31), thanks to the significant decrease in molecular weight (224 amu). In addition, the apparent permeability of 2 was greatly improved over 1 by 5-fold (14 × 10–6 cm/s, LLC-PK1), which was of particular importance for CNS penetration.
To further improve enzyme potency, insights from X-ray cocrystal structures and molecular modeling suggested that an ethyl group in the highly hydrophobic S2′ binding pocket was probably not optimal. Modifying 2 to better fill the S2′ pocket with larger hydrophobic substituents led to the neopentyl chromane derivative 3 as a potent BACE inhibitor (IC50 = 7.2 nM). Upon examination of modeled structures, we realized that the 8-position of the chromane moiety is open to a solvent-exposed area, suggesting that introduction of some polarity would not be detrimental to enzyme potency but could modulate physicochemical properties. We thus incorporated a nitrogen atom into the chromane at the 8-position. Gratifyingly, we found that such a subtle structural variation resulted in compound 4, which not only was equipotent as compound 3 in the enzyme assay but also displayed potent cellular activity (cell IC50, 3.1 nM). We next turned our attention to improving compound intrinsic stability in liver microsomes as a means to improve in vivo clearance in rodents. This communication will describe our efforts in detail on further optimization of 4, particularly modifications to the P1 substituent, leading ultimately to the discovery of 16 as a novel, potent, and orally bioavailable efficacious BACE inhibitor.
The synthesis of 4 and analogues 16–33 began with commercially available cyclobutanone (Scheme 1). The addition of allylmagnesium bromide to cyclobutanone, followed by protection of the secondary alcohol with tert-butyldimethylsilyl triflate, gave the silyl ether 5. Dihydroxylation was effected with osmium tetroxide and N-methylmorpholine-N-oxide to give diol 6, which was then treated with sodium periodate to afford aldehyde 7. The sulfinylimine 8 was formed by condensation with Ellman's chiral tert-butylsulfinamide auxiliary with copper sulfate.10 The fluoropyridine 9 was produced via a palladium-catalyzed zinc cross-coupling between commercially available reagents 2-fluoro-5-bromopyridine and neopentylzinc chloride. Fluoropyridine 9 was regioselectively deprotonated with lithium tetramethylpiperidide at the 3-position and then treated with sulfinylimine 8 to give the sulfinylamide adduct 10 in good yield with moderate diastereoselectivity (2:1) in favor of the desired diastereomer. Silyl ether deprotection of 10 with concurrent cyclization in the presence of cesium fluoride in DMSO at 120 °C furnished azachromane 11, which was readily separated from the minor diasteromer via column chromotagraphy. The chiral auxiliary was then smoothly cleaved under acidic conditions to give enantiomerically pure azachromane amine 12.
Scheme 1. Synthesis of Azachromane Intermediate 12.

Reagents and conditions: (a) Allylmagnesium bromide, THF, −78 °C. (b) TBSOTf, TEA, DCM, 0 °C. (c) OsO4, NMO, H2O:t-BuOH:THF (1:1:0.03). (d) NaIO4, THF:t-BuOH:H2O (1:1:2). (e) CuSO4, (R)-2-methylpropane-2-sulfinamide, DCM. (f) nBuLi, 2,2,6,6-tetramethylpiperidine, THF, −78 °C. (g) CsF, DMSO, 120 °C. (h) 4 M HCl, DCM.
We synthesized HEAs 2 and 3 (Figure 1) following well-established literature procedures that utilize a nucleophilic opening of an amino acid-derived epoxide with the appropriate chromane amine.5 We discovered that this methodology was sluggish and inefficient when an 8-azachromane amine was used as the nucleophile in this alkylation reaction. Several attempts to improve this epoxide opening with azachromane 12, including using Lewis acid additives, did not provide synthetically useful yields. Therefore, a reductive amination approach, one that circumvents the poor nucleophilic reactivity of 12, was developed to allow efficient coupling between the azachromane amine 12 with a suitable aldehyde 14 (Scheme 2).11 To complete the synthesis of inhibitors 4 and 16–32, the alcohol 13(12) was oxidized with Dess–Martin periodinane to give aldehyde 14. Sodium triacetoxyborohydride-promoted13 reductive amination gave a very smooth and highly efficient coupling reaction to provide adduct 15 in excellent yields. It is noteworthy that no epimerization of aldehyde 14 was observed in either the oxidation or the reductive amination sequence. Global deprotection of 15 was effected with HCl in methanol to give the penultimate hydroxyl diamine, which was then selectively acylated with acyl imidazole to give acetamides 4 and 16–32.
Scheme 2. Synthesis of BACE Inhibitors 16–32.

Reagents and conditions: (a) Dess–Martin periodinane, Na2CO3, DCM. (b) Compound 12, NaBH(OAc)3, trimethylorthoformate, DCM. (c) 4 M HCl, MeOH, 50 °C. (d) 1-(1H-Imidazol-1-yl)ethanone, DIPEA, DMF.
Metabolic identification studies of 4 in the presence of rat liver microsomes revealed that oxidation on the P1 ring and P2′ side chain was the major sites of oxidative metabolism. We surmised that the poor metabolic stability was one of the sources of high in vivo clearance (rat iv, 2 mg/kg, 5.3 L/h/kg). We explored modifications on the P1 aryl ring to address this metabolic soft spot of inhibitor 4. The results are summarized in Table 1.
Table 1. SAR of Phenyl Ring Substituentsa.
| μL/min/mgd |
|||||||
|---|---|---|---|---|---|---|---|
| compdb | R1 | R2 | BACE IC50 (nM)c | cell IC50 (nM)a | RLM | HLM | LLC-PK1 Papp (×10–6 cm/s) |
| 4 | H | H | 5.8 ± 6.3 | 3.1 ± 4.2 | 241 | 465 | 22 |
| 16 | 4-F | H | 5.0 ± 3.0 | 12 ± 6.1 | 180 | 107 | 21 |
| 17 | 3-F | H | 1.0 ± 0.47 | 1.8 ± 1.2 | 227 | 621 | 16 |
| 18 | 2-F | H | 3.6 ± 0.86 | 5.1 ± 4.3 | 206 | 342 | 13 |
| 19 | 4-Cl | H | 8.3 ± 2.7 | 13 ± 9.1 | 160 | 443 | 21 |
| 20 | 4-OCF3 | H | 14 ± 1.3 | 65 ± 4.3 | 101 | 181 | |
| 21 | 3-F | 5-F | 6.9 ± 8.7 | 6.6 ± 7.5 | 186 | 921 | 15 |
| 22 | 3-CF3 | H | 2.4 ± 0.97 | 4.0 ± 1.6 | 179 | 308 | 12 |
| 23 | 4-F | 2-F | 6.5 ± 5.3 | 18 ± 0.41 | 173 | 71 | |
| 24 | 4-F | 3-F | 2.0 | 2.4 ± 1.1 | 16 | ||
| 25 | 4-F | 3-CH3 | 4.0 ± 3.0 | 4.6 ± 2.6 | >399 | 829 | 15 |
| 26 | 4-F | 3-CF3 | 11 ± 14 | 18 ± 8.8 | 116 | 134 | 12 |
| 27 | 4-F | 3-OCF3 | 2.1 | 26 ± 18 | 220 | 360 | 8.6 |
| 28 | 4-F | 3-CN | 2.4 ± 0.06 | 4.5 ± 1.1 | 185 | 188 | |
| 29 | 4-F | 3-OCH3 | 4.5 ± 0.99 | 7.6 ± 3.4 | 165 | 628 | 20 |
| 30 | 4-CH3 | 3-F | 0.99 ± 0.37 | 2.2 ± 0.82 | 155 | >399 | 18 |
| 31 | 4-OCH3 | 3-F | 3.6 ± 0.73 | 8.5 ± 6.4 | 258 | 162 | 28 |
| 32c | 4-CF3 | 3-F | 44 ± 11 | 160 | 121 | 443 | 16 |
All values lacking standard deviations were measured as single data points.
All compounds were >95% pure by HPLC and characterized by 1H NMR and HRMS.
Compound is mixture of diastereomers (1:1, 2S,3R and 2R,3S).
Compounds are incubated with microsomes for 30 min at a concentration of 1 μM.
In general, we found that a wide range of substituents on the P1 aryl ring were well tolerated in terms of potency against the BACE enzyme. The first modification was to substitute a hydrogen atom with a single fluorine atom at different positions around the aryl ring. Placing a fluorine atom at the 4-position of the aryl ring (16) provided good potency and permeability. Moving the fluorine to the meta position (17) improved the potency 5-fold but was detrimental to stability in human liver microsomes (621 μL/mL/min). Moving the fluorine to the 2-position (18) provided good potency, but this compound was also not stable in human liver microsomes (342 μL/mL/min). Several alternative substititions were tried at the 4-position of the aryl ring, such as chlorine and trifluoromethoxy (19 and 20), but both were inferior to fluorine in terms of potency and metabolic stability. Substituting at both the 3- and the 5-positions with fluorine atoms (21) was consistent with single substitution with good potency but poor metabolic stability. Substituting trifluoromethyl for flourine (22) in the 3-position of the P1 phenyl group provided improved potency over 16 but suffered from poor human microsomal stability and reduced permeability. Placing fluorine atoms at both the 2- and the 4-positions (23) resulted in modest enzyme potency, but the increased lipohpilicity led to a larger enzyme to cell shift. Realizing that fluorine at the 4-position provided some stability and that the 3-position provided some improved potency, various substituents were tried at the 3-position in combination with the 4-fluoro substituent (24–29). The compounds were all comparable to 16 in potency, but almost all demonstrated poor stability in human liver microsomes. Other substituents at the 4-position (30–32) with fluorine at the 3-position generally provided good potency but failed to improve the metabolic stability of the molecules. These substituted P1 analogues have good potency and good permeability but generally suffer from poor in vitro microsomal stability (rat and human).
On the basis of favorable permeability and moderate in vitro microsomal stability, 16 was selected for further profiling. Pharmacokinetic analysis of 16 showed that in vivo clearance in rat was 1.2 L/h/kg, Vdss was 1.6 L/kg, and T1/2 was 7.2 h following an iv dose (2 mg/kg). Oral bioavailability was measured at 40% based on a 10 mg/kg dose.
Compound 16 was evaluated in a rat pharmacodynamic (PD) model. The model allowed for the measurement of acute reductions in soluble Aβ40 in plasma, brain, and CSF. The compound was administered at 100 mg/kg orally as a suspension in 2% HPMC, 1% Tween-80 at pH 2.0 to wild-type male Sprague–Dawley rats. Samples were collected from plasma, brain, and CSF 4 h after dosing.14 Compound 16 showed Aβ40 level reductions of 71, 32, and 35% in plasma, brain, and CSF, respectively (Figures 2–4). Concentrations of 16 at this time point were measured at 4.3, 0.66, and 0.006 μM, respectively, for the plasma, brain, and CSF. On the basis of the rat plasma protein binding (98.5%) of 16, the estimated unbound concentration was 64 nM in plasma and 10 nM in brain. The estimated unbound brain concentration is similar to the measured CSF concentration. The reduction in Aβ40 levels was consistent with the exposure levels and the in vitro cellular IC50.
Figure 2.

PD study of 16 in rat, plasma Aβ40 concentration (100 mg/kg po, 4 h postdose, P < 0.05). A literature γ-secretase inhibitor15 was used as a positive control (10 mg/kg, po).
Figure 4.

PD study of 16 in rat, CSF Aβ40 concentration (100 mg/kg po, 4 h postdose, P < 0.05). A literature γ-secretase inhibitor15 was used as a positive control (10 mg/kg, po).
Figure 3.

PD study of 16 in rat, brain Aβ40 concentration (100 mg/kg po, 4 h postdose, P < 0.05). A literature γ-secretase inhibitor15 was used as a positive control (10 mg/kg, po).
The binding of mode of compound 16 was determined by X-ray crystallography and is depicted in Figure 5.16 The hydroxyl group and basic amine of the HEA core engage in the expected contacts with the two catalytic aspartyl residues Asp32 and Asp228, respectively. The latter amine–Asp interaction is augmented by a hydrogen bond between the amine and the carbonyl oxygen of Gly34 (not shown.) Hydrogen-bonding interactions involving the acetamide moiety of the inhibitor and both the underside C=O of Gly230 and the backbone NH of flap Thr72, along with a CH···O type contact (ca. 3.4 Å) between C5 of the azachromane group and the C=O of Gly34, round out the additional, anchoring polar contacts between the inhibitor and the active site of BACE. The 4-fluoro substituent on the P1 aryl ring, although likely experiencing a somewhat repulsive interaction with the backbone carbonyl of Phe108, interacts with the edge of Trp115 and also likely helps to augment the attractive, edge-to-face interaction between the fluorophenyl substituent and the electron-rich π face of flap Tyr71. Interactions between the azachromane with Tyr198 (ca. 4.1 Å; not marked), occupation of S1′ by the spirocyclobutyl moiety, and occupation of the large S2′ cavity by the hydrophobic neopentyl group provide additional contacts contributing to the high binding affinity of 16.
Figure 5.
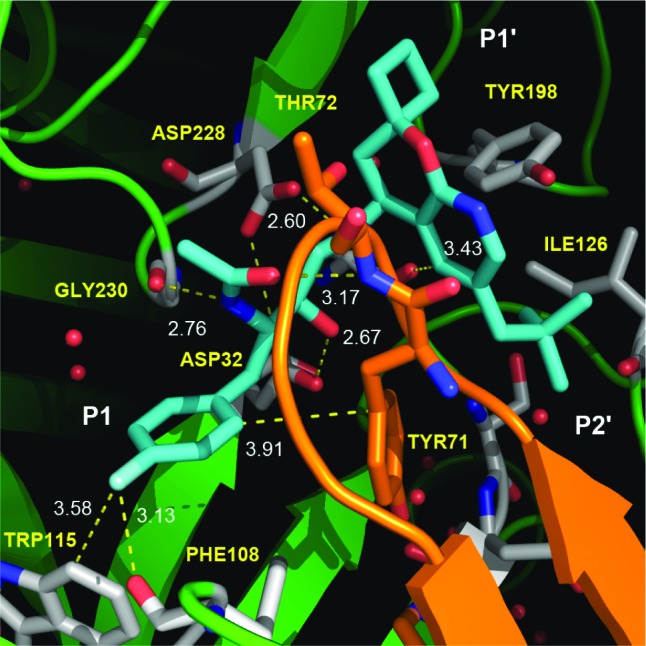
Crystal structure of inhibitor 16 bound to the active site of BACE.
In summary, starting from 1, we have improved several key parameters to help increase the likelihood of achieving CNS penetration. Compound 16 has a lower molecular weight (163 amu), fewer hydrogen bond donors and acceptors, and a lower PSA (from 105 to 83 Å2) as compared to inhibitior 1. We have explored the S1 pocket of BACE with several analogues, with compound 16 having the best profile. Oral administration of inhibitor 16 in a rat PD model demonstrated statistically significant lowering of Aβ40–42 peptide.
Acknowledgments
We gratefully acknowledge the Advanced Light Source. The Advanced Light Source is supported by the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Supporting Information Available
Procedures and characterization for all newly prepared compounds, coordinates for the cocrystal structure of inhibitor 16 with BACE, and experimental procedures for the enzyme and cellular assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Present Address
∇ Constellation Pharmaceuticals, 215 First Street, Suite 200, Cambridge, Massachusetts 02142, United States.
Author Present Address
○ Nimbus Discovery, 25 First Street, Suite 303, Cambridge, Massachusetts 02141, United States.
Author Present Address
¶ Envoy Therapeutics, 555 Heritage Drive, Jupiter, Florida 33458, United States.
Author Present Address
+ Sanofi-Aventis, 270 Albany Street, Cambridge, Massachusetts 02139, United States.
Author Present Address
¥ Eli Lilly and Company, Lilly Corporate Center, Indianapolis, Indiana 46285, United States.
Author Status
∞ Deceased.
Supplementary Material
References
- http://www.alz.org/documents_custom/report_alzfactsfigures2010.pdf.
- Vassar R.; Bennett B. D.; Babu-Khan S.; Kahn S.; Mendiaz E. A.; Denis P.; Teplow D. B.; Ross S.; Amarante P.; Loeloff R.; Luo Y.; Fisher S.; Fuller J.; Edenson S.; Lile J.; Jarosinski M. A.; Biere A. L.; Curran E.; Burgess T.; Louis J. C.; Collins F.; Treanor J.; Rogers G.; Citron M. β-Secretase Cleavage of Alzheimer's Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science 1999, 286, 735–741. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Tanzi R. E.; Bertram L. Twenty Years of the Alzheimer's Disease Amyloid Hypothesis: A Genetic Perspective. Cell 2005, 120, 545–555. [DOI] [PubMed] [Google Scholar]
- De Strooper B.; Vasar R.; Golde T The secretases: Enzymes with therapeutic potential in Alzheimer's disease. Nature Rev. Neurol. 2010, 6, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri R. Boom in the development of non-peptidic beta-secretase (BACE1) inhibitors for the treatment of Alzheimer's disease. Med. Res. Rev. 2008, 29, 295–338. [DOI] [PubMed] [Google Scholar]
- Stachel S. J. Progress toward the development of a viable BACE-1 inhibitor. Drug Dev. Res. 2009, 70, 101–110. [Google Scholar]
- Lopez P.; Zhong W.; Albrecht B.; Amarante P.; Andersen D.; Babu-Khan S.; Bartberger M. D.; Brown J.; Chen K.; Cheng Y.; Citron M.; Croghan M. D.; Graceffa R.; Harried S. S.; Hickman D.; Hungate R.; Jordan S.; Kaller M. R.; Kriemen C.; La D.; Luo Y.; San Miguel T.; Monenschein H.; Nguyen T.; Nixey T.; Pennington L. D.; Wahl R. C.; Weiss M. M.; Wood S.; Xue M.; Yang B.; Yan Q.; Zhang J; Zhang J.; Patel V.; Hitchcock S.. Optimization of P1′ Interactions in a Series of Hydroxyethylamine Derived Small Molecule BACE1 Inhibitors. Manuscript in preparation.
- Lanz T. A.; Himes C. S.; Pallante G.; Adams L.; Yamazaki S.; Amore B.; Merchant K. M. The γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester reduces Abeta levels in vivo in plasma and cerebrospinal fluid in young (plaque-free) and aged (plaque-bearing) Tg2576 mice. J. Pharmacol. Exp. Ther. 2003, 305, 864–871. [DOI] [PubMed] [Google Scholar]
- Reynolds C. H.; Tounge B. A.; Bembenek S. D. Ligand Binding Efficiency: Trends, Physical Basis, and Implications. J. Med. Chem. 2008, 51, 2432–2438and references therein. [DOI] [PubMed] [Google Scholar]
- Hitchcock S. A.; Pennington L. D. Structure-Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [DOI] [PubMed] [Google Scholar]
- Maillard M.; Baldwin E. T.; Beck J. T.; Hughes R. T.; Varghese J.; Pulley S. R.; Tenbrink R.. Acetyl 2-Hydroxy-1,3-diaminoalkanes. PCT Int. Appl. WO 2004024081, 2004.
- Ellman J. A.; Owens T. D.; Tang T. P. N-tert-Butanesulfinyl Imines: Versatile Intermediates for the Asymmetric Synthesis of Amines. Acc. Chem. Res. 2002, 35, 984–995. [DOI] [PubMed] [Google Scholar]
- Parkes K. E. B.; Bushnell D. J.; Crackett P. H.; Dunsdon S. J.; Freeman A. C.; Gunn M. P.; Hopkins R. A.; Lambert R. W.; Martin J. A.; Merret J. H.; Redshaw S.; Spurden W. C.; Thomas G. J. Studies toward the large-scale synthesis of the HIV proteinase inhibitor Ro 31-8959. J. Org. Chem. 1994, 59, 3656–3664. [Google Scholar]
- Harried S. S.; Croghan M. D.; Kaller M. R.; Lopez P.; Zhong W.; Hungate R.; Reider P. J. Stereoselective Synthesis of anti-N-Protected 3-Amino-1,2-epoxides by Nucleophilic Addition to N-tert-Butanesulfinyl Imine of a Glyceraldehyde Synthon. J. Org. Chem. 2009, 74, 5975–5982. [DOI] [PubMed] [Google Scholar]
- Abdel-Magid A. F.; Carson K. G.; Harris B. D.; Maryanoff C. A.; Shah R. D. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures. J. Org. Chem. 1996, 61, 3849–3862. [DOI] [PubMed] [Google Scholar]
- Drug levels were quantified using the standard LC/MS/MS methods. Aβ40 levels were quantified using ELISA-based Meso Scale Discovery (MSD) technology.
- Example 7-B [(S)-2-hydroxy-3-methyl-N-((S)-1-(((S)-5-methyl-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl)amino)-1-oxopropan-2-yl)butanamide]. Wu J.; Tung J. S.; Thorsett E. D.; Pleiss M. A.; Nissen J. S.; Neitz J.; Latimer L. H.; Varghese J.; Freedman S.; Britton T. C.; Audia J. E.; Reel J. K.; Mabry T. E.; Dressman B. A.; Cwi C. L.; Droste J. J.; Henry S. S.; McDaniel S. L.; Scott W. L.; Stucky R. D.; Porter W. J.. Cycloalkyl, lactam, lactone and related compounds, pharmaceutical compositions comprising same, and methods for inhibiting β-amyloid peptide release and/or its synthesis by use of such compounds. International Patent Application WO 98/28268, 1998.
- Coordinates for the complex of BACE with inhibitor 16 have been deposited in the Protein Data Bank under PDB ID code 4DUS.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.